Case Studies in Structure-Based Design¶
Info
Successful examples of structure-based drug design projects are outlined in this chapter with a focus on the drug design features, in particular the analyses that were made and how they were exploited for the successful design of active compounds.
Number of Pages: 75 (±1 hours read)
Last Modified: December 2008
Prerequisites: Principles of Rational Drug Design, Structure-Based Drug Design: Analysis, Structure-Based Drug Design: Design
Case Study-1 : Phenyl Imidazoles¶
Phenyl-Imidazoles Inhibit Cytochrome P450¶
Cytochrome P450 is a group of enzymes which contain Heme and catalyze the hydroxylation of aliphatic and aromatic molecules. Camphor is the natural substrate of the bacterial cytochrome P450-cam. Phenylimidazoles, such as phenyl-1, phenyl-2 or phenyl-4, are known to inhibit this hydroxylation process. From a structural point of view a simple rationale could be derived to explain these observations.

articles
Crystal Structures of Metyrapone- and Phenylimidazole-Inhibited Complexes of Cytochrome P-450cam Poulos TL and Howard AJ Biochemistry 26 1987
High-Resolution Crystal Structure of Cytochrome P450cam Poulos TL, Finzel BC, and Howard AJ J Mol Biol. 195 1987
Simple Consideration: Shape Similarity¶
It is reasonable to assume that the three inhibitors bind to the enzyme in a way where all the phenyl rings are aligned. This superimposition has the advantage of showing that the three molecules have almost identical shapes and can therefore occupy the same hypothetical sub-pocket in the enzyme catalytic site.

Perhaps Binding Elements are more Complex ?¶
However, is the molecular shape the only molecular determinant for binding? Perhaps electrostatic interactions or anchorage with hydrogen bonds are also important! Thus the previous model which considered the geometrical features alone (and does not distinguish between nitrogen and carbon atoms) is not sufficient. To satisfy the alignment of electronic features, we need to consider aligning the molecules in such a way that the nitrogen atoms are optimally superimposed. However the resulting model is far from being convincing.

The Structure-Based Answer¶
In the absence of additional information, we would probably tend to favor the initial pure steric model. In fact additional information is available, as the X-ray structures of the complexes have been solved, including the three phenyl-imidazole inhibitors, the natural substrate camphor and other potent inhibitors. By aligning the proteins of the complexes, the exact solution how the three phenyl-imidazole analogs should be superimposed is now accessible. One important element in the binding of these molecules is their chelation between one nitrogen atom and the iron of the P450 protein. This is shown in the browser on the next page.

articles
Crystal Structures of Metyrapone- and Phenylimidazole-Inhibited Complexes of Cytochrome P-450cam Poulos TL and Howard AJ Biochemistry 26 1987
Phenyl-Imidazole Browser¶

articles
Crystal Structures of Metyrapone- and Phenylimidazole-Inhibited Complexes of Cytochrome P-450cam Poulos TL and Howard AJ Biochemistry 26 1987
Limitations of Chemical Intuition¶
Although in many cases chemical intuition can compensate for lack of knowledge, there are situations where in the absence essential data, counterproductive models are developed. This example illustrates this situation particularly well and shows the strength of structure-based analyses.

Case Study-2 : BACE-1 Inhibitors¶
BACE-1 Inhibitors¶
In order to discover therapeutic agents for treating the etiology of Alzheimer's disease, Johnson & Johnson addressed the inhibition of the aspartic protease BACE-1 (β-Site APP Cleaving Enzyme), based on the observation that the inhibition of this enzyme lowers β-amyloid levels in the brains of mice. A structure-based strategy was used, which is presented in the following pages.

articles
The novel beta-secretase inhibitor KMI-429 reduces amyloid beta peptide production in amyloid precursor protein transgenic and wild-type mice Asai M, Hattori C, Iwata N, Saido TC, Sasagawa N, Szabo B, Hashimoto Y, Maruyama K, Tanuma S, Kiso Y, Ishiura S. J Neurochem 96(2) 2006
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
Screening the J&J Corporate Compound Collection¶
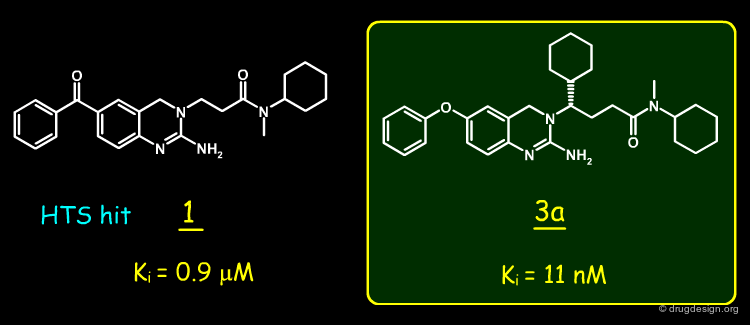
A random screening of the J&J corporate compound collection led to the identification of a hit such as the 2-amino-3,4-dihydroquinazoline 1, having a Ki of 0.9 µM. The compound was selected for further studies.
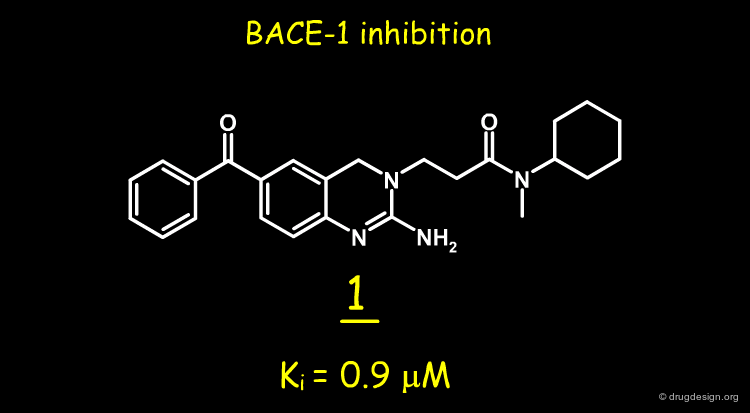
articles
2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (beta-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead Ellen W. Baxter, Kelly A. Conway, Ludo Kennis, Francois Bischoff, Marc H. Mercken, Hans L. De Winter, Charles H. Reynolds, Brett A. Tounge, Chi Luo, Malcolm K. Scott, Yifang Huang, Mirielle Braeken, Serge M. A. Pieters, Didier J. C. Berthelot, Stefan Masure, Wouter D. Bruinzeel, Alfonzo D. Jordan, Michael H. Parker, Robert E. Boyd, Junya Qu, Richard S. Alexander, Douglas E. Brenneman, and Allen B. Reitz J.Med.Chem 50 2007
Structural Determinants of the Biological Activity of 1¶
Nothing was known about the structural determinants responsible for the inhibitory activity of compound 1. The J&J group was interested in finding out how the hit compound 1 interacts with BACE-1, to take advantage of such knowledge to rationally develop more potent inhibitors. In the framework of collaboration with Astex Therapeutics, the X-ray structure of BACE-1 complexed with compound 1 was solved.
articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
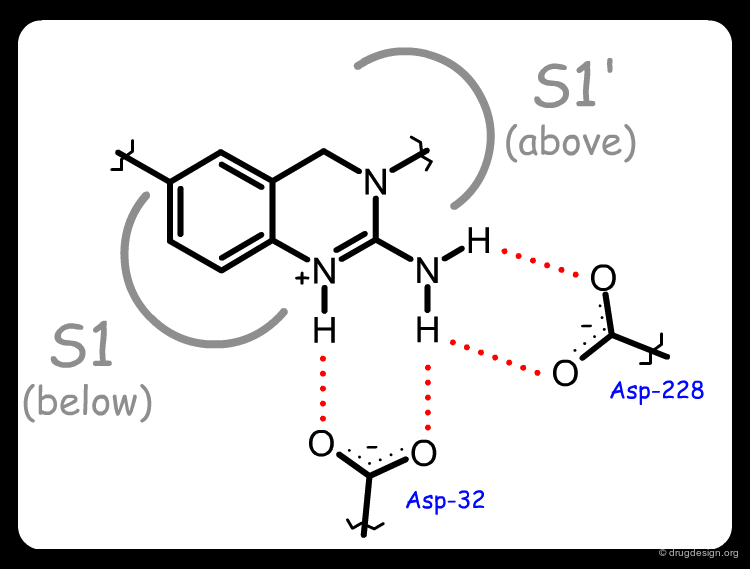
X-ray Structure of the Complex of 1 with BACE-1¶
The X-ray structure of the complex of 1 with BACE-1 revealed several interesting features. Molecule 1 was found in an unexpected compact folded conformation, the side chain of the molecule bending back onto itself in a hairpin turn orientation enabling the N-cyclohexyl substituent to occupy the hydrophobic S1 pocket. An exquisite array of hydrogen bonds was formed between the ligand and the two aspartates of the catalytic site (see third button).


articles
2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (beta-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead Ellen W. Baxter, Kelly A. Conway, Ludo Kennis, Francois Bischoff, Marc H. Mercken, Hans L. De Winter, Charles H. Reynolds, Brett A. Tounge, Chi Luo, Malcolm K. Scott, Yifang Huang, Mirielle Braeken, Serge M. A. Pieters, Didier J. C. Berthelot, Stefan Masure, Wouter D. Bruinzeel, Alfonzo D. Jordan, Michael H. Parker, Robert E. Boyd, Junya Qu, Richard S. Alexander, Douglas E. Brenneman, and Allen B. Reitz J.Med.Chem 50 2007
Flap Flexibility in Aspartyl Proteases¶
It was also found that the BACE-1 flap (that is located over the active site of the enzyme), was pushed away by a distance of about 4 Å when compared to the structure of the peptidic inhibitor OM99-2. This type of flexibility was also observed for the flap region of other aspartyl proteases such as HIV-1 and Renin.

articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
Compound with Increased Folding Capability¶
The J&J team looked for molecules that mimic the folded conformation of molecule 1 and designed compound 2. The molecule had a longer side-chain and could more easily adopt a hairpin conformation. Also for synthetic reasons the carbonyl of the benzoyl group present in 1 was replaced by an oxygen. This molecule was synthesized and proved to have a Ki of 158 nM.


articles
2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (beta-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead Ellen W. Baxter, Kelly A. Conway, Ludo Kennis, Francois Bischoff, Marc H. Mercken, Hans L. De Winter, Charles H. Reynolds, Brett A. Tounge, Chi Luo, Malcolm K. Scott, Yifang Huang, Mirielle Braeken, Serge M. A. Pieters, Didier J. C. Berthelot, Stefan Masure, Wouter D. Bruinzeel, Alfonzo D. Jordan, Michael H. Parker, Robert E. Boyd, Junya Qu, Richard S. Alexander, Douglas E. Brenneman, and Allen B. Reitz J.Med.Chem 50 2007
How to Gain Additional Binding¶
Encouraged by the results obtained on compound 2 and the observation that the hydrophobic S1' pocket of the enzyme was not occupied by the inhibitors, the J&J team looked for molecules that could form favorable interactions with this pocket. This led to considering substituents having the S stereochemistry on the α-carbon of the side chain of the inhibitors and helped show that a cyclohexyl group fit perfectly this pocket perfectly.

articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
Design of a More Potent Inhibitor¶
This idea led to the design of 3, which proved to be active at the nanomolar level. The enantiomers of 3 were prepared and the biological activity was found to reside predominantly in the S enantiomer (3a) as predicted.

articles
2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (beta-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead Ellen W. Baxter, Kelly A. Conway, Ludo Kennis, Francois Bischoff, Marc H. Mercken, Hans L. De Winter, Charles H. Reynolds, Brett A. Tounge, Chi Luo, Malcolm K. Scott, Yifang Huang, Mirielle Braeken, Serge M. A. Pieters, Didier J. C. Berthelot, Stefan Masure, Wouter D. Bruinzeel, Alfonzo D. Jordan, Michael H. Parker, Robert E. Boyd, Junya Qu, Richard S. Alexander, Douglas E. Brenneman, and Allen B. Reitz J.Med.Chem 50 2007
X-Ray Structure of the Complex with 3a¶
The complex of BACE-1 with 3a was solved and confirmed the predicted binding mode of molecule 3a: the occupancy of the S1' pocket by the newly introduced cyclohexyl, a folded conformation of the ligand that put the cyclohexyl group of the side-chain in the S1 pocket, and the network of hydrogen-bond interactions between the N-H groups of the ligand with the two aspartates of the catalytic site of the enzyme.


articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
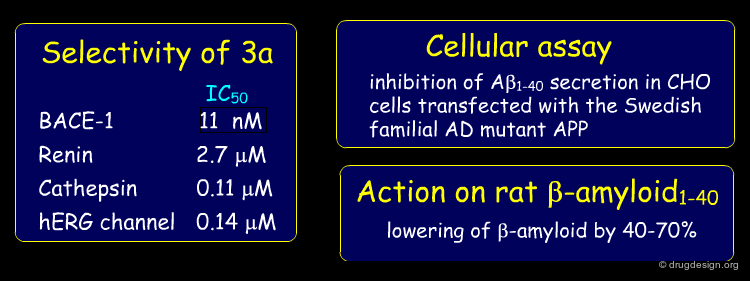
Pharmacological Action of Compound 3a¶
Compound 3a was further examined to assess its suitability as a lead. The compound showed some selectivity with respect of other aspartic proteases; it exhibited good potency in a cellular assay and proved to lower β-amyloid in rat plasma after p.o. administration.
articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
Important Structural Determinants for Binding¶
The J&J group successfully raised the potency of a weak inhibitor which was found by screening to the nano-molar level. Four important structural determinants were exploited in the design process and enabled the success of this phase of the project: the good anchorage of the 2-aminoquinazoline moiety to the aspartates of the catalytic site; the folded bioactive conformation adopted by the inhibitors enabling it to bind to the hydrophobic S1 pocket and the presence of a yet unexploited hydrophobic pocket in S1'.

articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
Summary¶
A structure-based strategy that exploited X-ray crystallography and the docking of new prototypes enabled the rapid discovery of potent inhibitors such as 3a (Ki = 11 nM). The molecule showed good pharmacological properties and is currently being optimized to target Alzheimer's disease. The synthetic effort would not have been given high priority without the insight provided by the crystal structure of the initial hit bound to BACE-1.
articles
Emerging beta-amyloid therapies for the treatment of Alzheimer's disease Conway KA, Baxter EW, Felsenstein KM, Reitz AB Curr Pharm Des 9(6) 2003
Case Study-3 : Factor Xa Inhibitors¶
Therapeutic Utility of Factor Xa Inhibitors¶
Antithrombotic drugs can be designed by targeting the inhibition of the coagulation cascade outlined below. It was suggested that inhibition of factor Xa would be more efficient than inhibiting thrombin for developing novel antithrombotic agents, on the basis of calculations that show that one molecule of factor Xa generates 138 molecules of thrombin. Here we present the development of a design strategy that led to the discovery of a new and potent Factor Xa inhibitor.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
DX-9065a : a Factor Xa Inhibitor¶
DX-9065a is a potent and selective factor Xa inhibitor with an IC50 of 41 nM.


articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Complex Between Factor Xa and DX-9065a¶
The crystal structure of DX-9065a bound to factor Xa has been resolved. It is visualized here with some details.



articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Analysis of the Factor Xa and DX-9065a Complex (1/4)¶
DX-9065a binds to the receptor site with the naphthyl-amidine hydrogen bonding to Asp-189 in the S1 site.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Analysis of the Factor Xa and DX-9065a Complex (2/4)¶
The carboxylic acid makes hydrogen bonds with Gln-192.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Analysis of the Factor Xa and DX-9065a Complex (3/4)¶
The acetimiodyl pyrrolidine group binds to the aryl binding pocket S4 in a cation hole created by the carbonyl groups of Lys-96 and Glu-97 together with the Glu-97 side-chain.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Analysis of the Factor Xa and DX-9065a Complex (4/4)¶
The interactions of DX-9065a when it binds to factor Xa are represented in the following diagram.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Role of the Carboxylic Acid in Selectivity (1/3)¶
The specificity of DX-9065a for binding to factor Xa rather than thrombin is thought to stem for the favorable interaction of the carboxylate with the Gln-192 side-chain of factor Xa.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Role of the Carboxylic Acid in Selectivity (2/3)¶
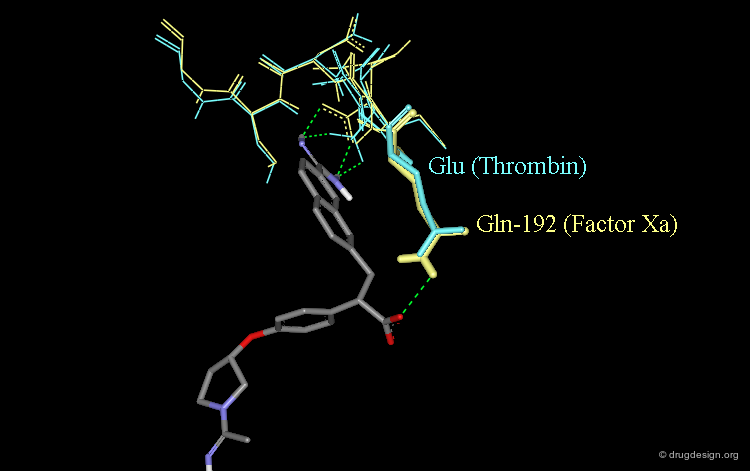
The corresponding residue in thrombin is Glu-192 which is thought to be deprotonated; this would cause a strong repulsive interaction with the carboxylate moiety of DX-9065a.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Role of the Carboxylic Acid in Selectivity (3/3)¶
The following view shows the interactions of DX-9065a with both factor Xa (yellow) and thrombin (blue).
articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Initial Inhibitor Design¶
The design idea envisioned two phenyl-amidine groups in a molecule with one of them binding in the S1 pocket, and the second in the S4 pocket.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Design (step 1): Structural Moiety for Pocket S1¶
It was assumed that the first phenyl-amidine group would enter the S1 pocket of factor Xa in a similar way as observed in the complex between trypsin and phenyl-amidine (shown below).

Phenyl-Amidine Entered into the S1 Pocket¶
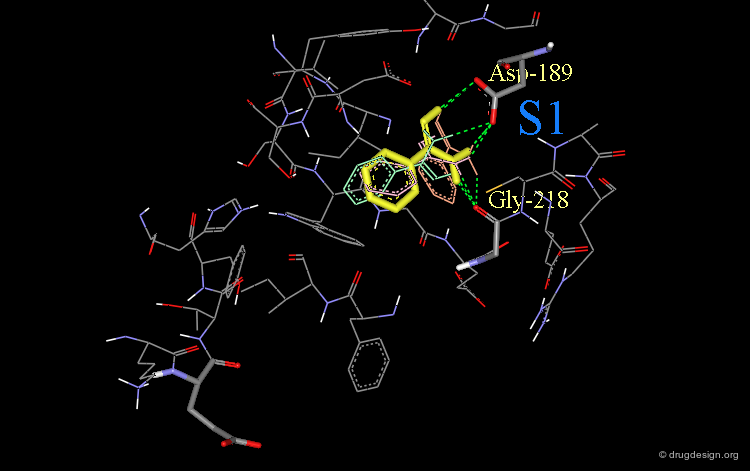
This phenyl-amidine group was therefore entered into the S1 pocket of factor Xa, in order to form hydrogen bonds with the side-chains of Asp-189 and Gly-218.

Phenyl-Amidine Oriented in Lowest Energy Orientation¶
Finally, the lowest energy orientation (in yellow) of the phenyl-amidine group was calculated in the S1 pocket.
articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Design (step 2): Structural Moiety for Pocket S4¶
The aryl-binding pocket S4 consists of three aromatic residues: Phe-174, Tyr-99 and Trp-215.

Phenyl Ring Introduced in Pocket S4¶
Initially, a phenyl ring was introduced in this pocket (S4) to find an optimal binding orientation.

Phenyl Substituted with an Amidine¶
The phenyl ring fragment was replaced with an amidine group, and the orientation of the phenyl-amidine moiety was then modeled to achieve optimal interactions in the S4 pocket.

Stacking Interaction of Phenyl-Amidine with Trp-215¶
Finally, an orientation was obtained where the amidine stacked over Trp-215 (3D)

Phenyl-Amidine Orientation¶
Note that the orientation of the phenyl-amidine is different from the position of the DX-9065a acetimidoyl group (in purple).

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Design (step 3): Design of the Spacer¶
Assuming the orientation of the phenyl-amidine groups in S1 and in S4 to be preferred, the design process consisted of connecting these fragments without disrupting their respective orientations.
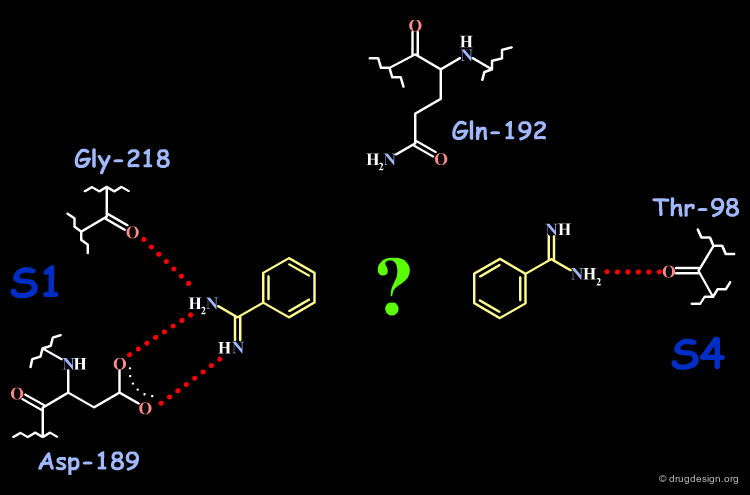
Phenyl-Amidine Groups in their Preferred Orientations¶
The following view shows the two phenyl-amidine groups in their preferred orientations.

Spacer with three Atoms¶
A 3-atom spacer was found connecting the meta position of the phenyl-amidine located in S1 and the para position of the phenyl-amidine group located in S4.

Candidate Prototype in the Catalytic Site¶
The two phenyl-amidino fragments and the 3-atom spacer are visualized here in the catalytic site of the enzyme.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Design (step 4): Positioning of the Carboxylate¶
Finally, the functionalization of the benzylic methylene of the phenyl-amidine group located in pocket S1 proved to be appropriate for introducing a carboxylate group to create favorable interactions with the side-chain of Gln-192, in a similar way as observed with the DX-9065a inhibitor.
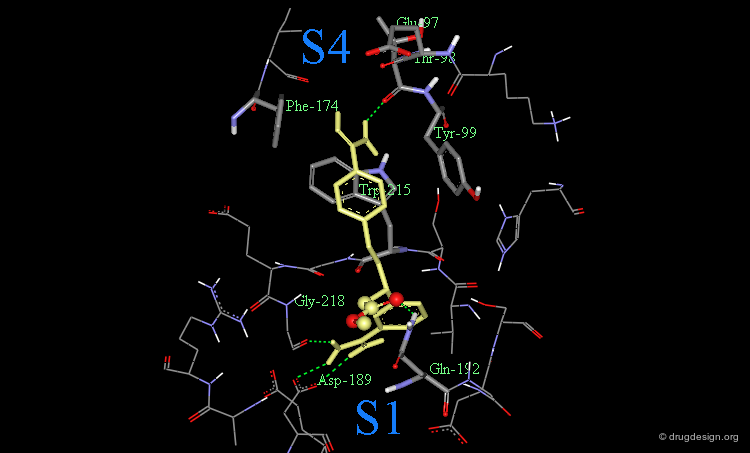
articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Discovery of a Lead Compound¶
The initial prototype compound shown below was synthesized and proved to have a good binding affinity for factor Xa with Ki = 34 nM.

articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Optimization of the Designed Series¶
The initial lead was further optimized. It was found that introducing a guanidine moiety (compound 21) significantly improved the inhibitory potency for Factor Xa (Ki = 9 nM) with a selectivity of about 10-fold as compared to thrombin.

Interaction of Compound 21 with Factor Xa¶
The following is a view of the interaction of compound 21 with factor Xa. Note that this guanidine analog fills the aryl-binding pocket more completely than the amidino group. It also extends deeper into the S4 pocket.

Finding an Optimal Spacer¶
Attempts to extend even deeper into S4 with a 5-atom spacer (compound 23) or more did not lead to better compounds. The preferred distance proved to be close to the 3-atom spacer.
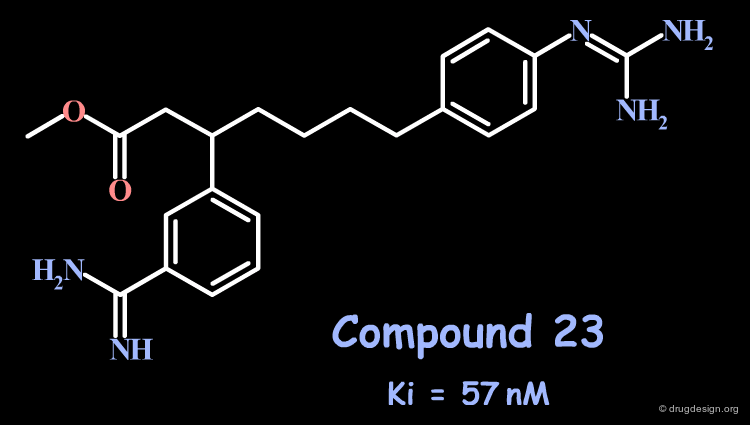
articles
Rational Design and Synthesis of Novel, Potent Bis-phenylamidine Carboxylate Factor Xa Inhibitors Maduskuie TP Jr., McNamara KJ, Ru Y, Knabb RM and Stouten PFW Journal of Medicinal Chemistry 41 1998
Case Study-4 : Kinase Inhibitors¶
Pyrrolo-Pyrimidine & Quinazoline EGF-R Inhibitors¶
In the process of R&D on inhibitors of the EGF-R protein kinase as anticancer agents, Novartis discovered the pyrrolo-pyrimidine scaffold while Parke-Davis identified quinazoline structures. In both cases, the molecules proved to be competitive inhibitors of the ATP substrate, although the models used were different. In the following pages, we discuss the structure-based analyses that were made by the two groups to prove the validity of their respective models.


articles
Modelling Study Of Protein Kinase Inhibitors: Binding Mode Of Staurosporine and Origin Of The Selectivity Of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U and Traxler P J. Comput. Aided. Mol. Des. 6 1995
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
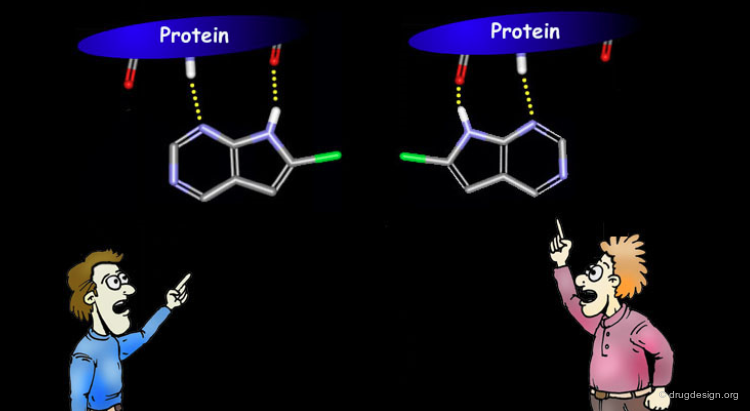
Novartis and Parke-Davis Opposite Binding Models¶
This account is interesting: not only because each team succeeded in finding potent inhibitors in their own series, but also because of the unresolved disagreement.
articles
Modelling Study Of Protein Kinase Inhibitors: Binding Mode Of Staurosporine and Origin Of The Selectivity Of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U and Traxler P J. Comput. Aided. Mol. Des. 6 1995
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
Controversy: Novartis & Parke-Davis Binding Modes¶
The disagreement relates to how the molecules are recognized by the protein. The view shown here summarizes the binding mode imagined by each group for their own molecules.

Parke-Davis Analyses¶
The following view illustrates the binding mode used by Parke-Davis for its own series (left) and how they viewed the binding of the Novartis molecules (right).

Novartis Analyses¶
The following view illustrates the binding mode used by Novartis for its own series. It was shown on the previous page that two alternative binding modes can be imagined for the Novartis molecule, however only one binding mode can apparently be modeled for the Parke-Davis compound.

articles
Modelling Study Of Protein Kinase Inhibitors: Binding Mode Of Staurosporine and Origin Of The Selectivity Of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U and Traxler P J. Comput. Aided. Mol. Des. 6 1995
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
X-ray Structure of ATP Bound to a Kinase¶
The Rational Drug Design Strategies¶
Lead compounds were discovered through pharmacophore-based strategies. However, the crystal structure of a cyclic-AMP dependent kinase complexed with ATP, the cofactor of the protein kinases, revealed the key hydrogen bonds exploited by ATP in its binding to the kinase. This first crystallographic structure opened the door to a structure-based design perspective and facilitated the optimization of the leads.

articles
Crystal Structure of the Catalytic Subunit of cAMP-Dependent Protein Kinase Complexed with MgATP and Peptide Inhibitor Zheng J, Knighton DR, ten Eyck LF, Karlsson R, Xuong N, Taylor SS, and Sowadski JM Biochemistry 32 1993
Binding Mode of ATP¶
ATP, the cofactor of the protein kinases was found to anchor to the kinases in a bidentate manner with backbone atoms of two residues of the enzyme. This anchorage, reminiscent of the Watson-Crick mode of base pairing of adenine nucleotides, appeared to be a key determinant in the recognition of ATP by the kinases.

Binding Mode of Staurosporine¶
Staurosporine is an ATP competitive potent protein kinase inhibitor. Despite its seemingly unrelated chemical structure it was shown that the compound could exploit the same hydrogen bond interactions as ATP.

articles
Modelling Study Of Protein Kinase Inhibitors: Binding Mode Of Staurosporine and Origin Of The Selectivity Of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U and Traxler P J. Comput. Aided. Mol. Des. 6 1995
Homology Model of EGF-R Catalytic Site¶
The X-ray data also served in the construction of a model of the EGF-R kinase by homology and the identification of different areas in the active site (the anchorage, the "sugar" pocket, and the "large" pocket).

From Staurosporine to Pyrrolo-pyrimidine¶
The Novartis predicted binding mode for Staurosporine proved to be correct as demonstrated by subsequent X-ray studies of complexes with other protein kinases. Novartis ascribed a binding mode exploiting the same hydrogen bond interactions to the pyrrolo-pyrimidine series.

articles
Modelling Study Of Protein Kinase Inhibitors: Binding Mode Of Staurosporine and Origin Of The Selectivity Of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U and Traxler P J. Comput. Aided. Mol. Des. 6 1995
The Novartis Binding Mode of Pyrrolo-pyrimidine¶
The Novartis group placed the anilino moiety in the relative small region defined as the "sugar" pocket because the biological activities were very sensitive to the substitution in the phenyl ring. For example only a para-fluoro substitution is accepted as opposed to all other analogs with a substitutions in para, (e.g. with a chlorine, a methyl or ethyl) which were inactive.

articles
Modelling Study Of Protein Kinase Inhibitors: Binding Mode Of Staurosporine and Origin Of The Selectivity Of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U and Traxler P J. Comput. Aided. Mol. Des. 6 1995
The Pyrrole Ring in the Large Pocket¶
The pyrrole ring was placed in the "large" pocket area because substitutions of the heterocycle by many hydrophobic groups improved the biological activities. The analogs with two methyls, one methyl and one phenyl or two additional phenyl rings substantially increased biological activities as shown below.

The Pyrrole Ring Pointing Towards the Sugar Pocket¶
The "reverse" binding mode, with the pyrrole ring pointing towards the sugar pocket, is not a valid model. It eliminates the possibility of substitutions on the pyrrole ring. Such analogs were actually synthesized and increased the biological activities, which could not be accounted for by this model.

Parke-Davis Analyses the Quinazoline Scaffold¶
The rationale behind the Parke-Davis group's idea for modeling the binding mode of their quinazoline scaffold was based on systematic exploration of the anchorage capabilities of this scaffold. The two nitrogens of the quinazoline nucleus were shown to be essential for these activities (inactive compounds are obtained when any of the nitrogens is replaced by a carbon). The Parke-Davis group concluded that both atoms should be used as hydrogen bond acceptors in the interaction with the enzyme.

Additional SAR Analyses made by Parke-Davis¶
Substitution of the phenyl ring of the anilino side chain greatly reduced activities indicating limited bulk tolerance, and only the analog with bromine in meta showed improved activities.

articles
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
Parke-Davis Model of the Quinazoline Analogs¶
All the elements assembled by the Parke-Davis group lead to considering the following binding mode for the quinazoline series.

Specificity Observed in EGF-R Kinase Inhibition¶
An additional element that supported this model was the specificity observed in EGF-R activities. This can be explained by favorable hydrophobic interactions with Cys-751 that exist only in the EGF-R enzyme.

Anilino Towards the Sugar Pocket not Reasonable¶
The "reverse" binding mode, with the anilino side chain pointing towards the sugar pocket, is a model that does not explain the observed selectivity.
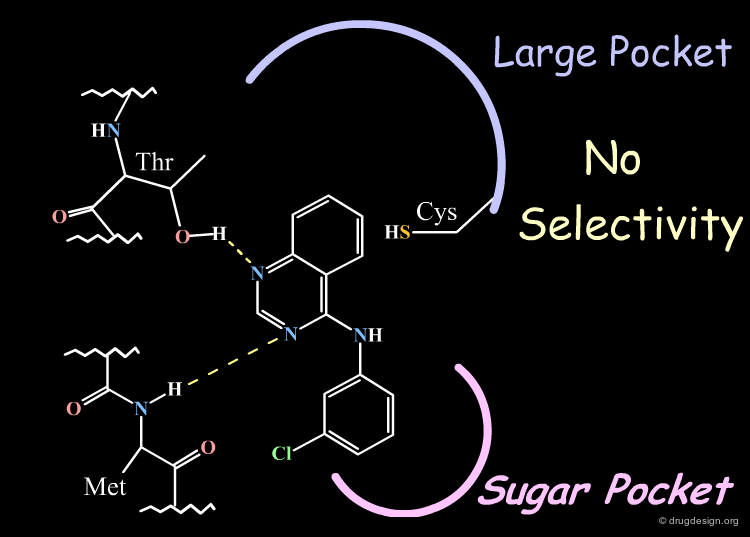
articles
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
Parke-Davis Model Consistent with Observed SAR¶
This binding mode indicates that the available room around the nitrogen area is very limited, whereas a third ring can be fused to the bicyclic quinazoline ring without loss of binding affinity. Docking studies showed that the binding mode represented here satisfied all the observed SAR.
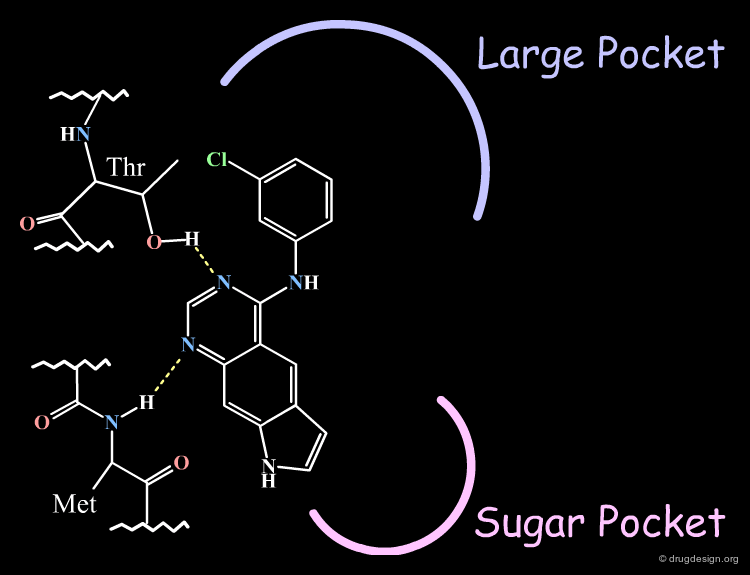
articles
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
Binding Mode of the Pyrrolo-Pyrimidine Series¶
The following example illustrates the binding mode of pyrrolo-pyrimidine as imagined by Novartis or Parke-Davis.
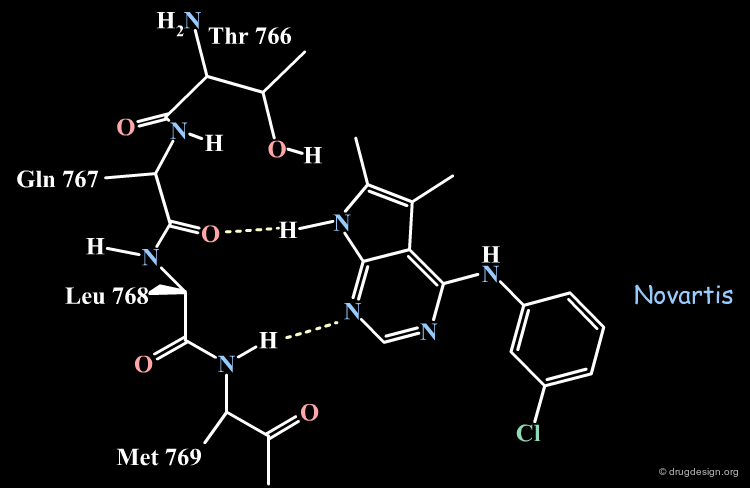
articles
Use of a Pharmacophore Model for the Design of EGFr Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4,-d]pyrimidines Trumpp-Kollmeyer S and Showalter HDH Chemtracts - Org. Chem. 11 1998
Binding Mode of the Quinazoline Series¶
The following example illustrates the binding mode of the quinazoline as imagined by Novartis or Parke-Davis.


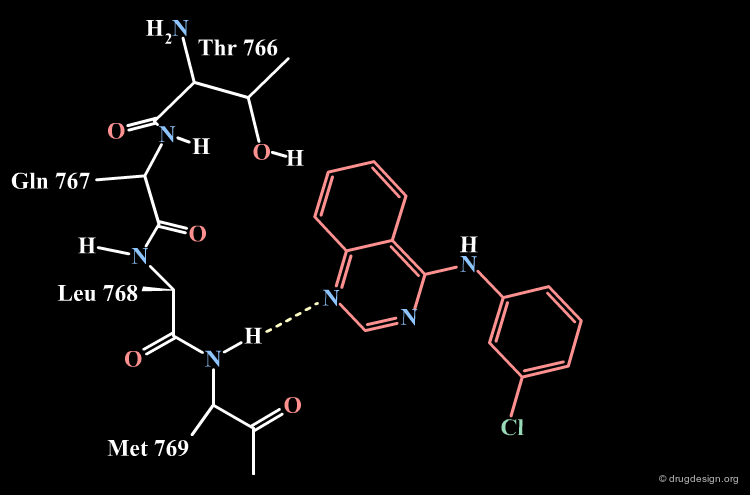
What is the Correct Solution?¶
The dispute is not yet resolved. In principle, there are three possibilities.

Ligand Observed with a Novartis Binding Mode¶
The following example illustrates the X-ray structure of a complex of NU2058 with another protein kinase, CDK2, revealing an orientation of the ligand in a Novartis-like binding mode.

articles
Identification of Novel Purine and Pyrimidine Cyclin-Dependent Kinase Inhibitors with Distinct Molecular Interactions and Tumor Cell Growth Inhibition Profiles Arris CE, Boyle FT, Calvert AH, Curtin NJ, Endicott JA, Garman EF, Gibson AE, Golding BT, Grant S, Griffin RJ, Jewsbury P, Johnson LN, Lawrie AM, Newell DR, Noble ME, Sausville EA, Schultz R and Yu W J. Med. Chem. 43 2000
Alignment with the Novartis Model¶
The Novartis model (yellow) is displayed aligned with the X-ray structure of the NU2058 bound to CDK2.
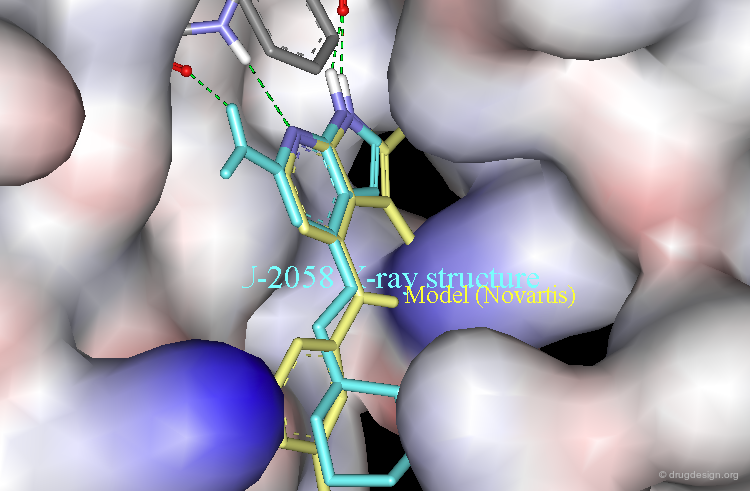
Ligand Observed with a Parke-Davis Binding Mode¶
The following example illustrates the X-ray structure of a complex of a 4-anilinoquinazoline derivative with the protein kinase p38, revealing an orientation of the ligand in a Parke-Davis-like binding mode.

articles
Binding Mode of the 4-Anilinoquinazoline Class of Protein Kinase Inhibitor: X-ray Crystallographic Studies of 4-Anilinoquinazolines Bound to Cyclin-Dependent Kinase 2 and p38 Kinase Shewchuk L, Hassell A, Wisely B, Rocque W, Holmes W, Veal J and Kuyper LF J. Med. Chem. 43 2000
Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor Stamos J, Sliwkowski MX, Eigenbrot C J Biol Chem. 277 2002
Alignment with the Parke-Davis Model¶
The Parke-Davis model (yellow) is displayed aligned with the X-ray structure of the 4-anilinoquinazoline bound to p38.

X-Ray Resolution of Tarceva Bound to EGF-R Kinase¶
Very recently, the crystal structure of the EGF-R kinase domain complexed with Tarceva (an inhibitor currently in development) has been determined. The orientation of the inhibitor in the complex corresponds to the Parke-Davis model and confirms the existence of a water molecule, as already observed in p38-inhibitor complexes.

Conclusion¶
Two opposite models were derived from analyses of activities on the EGF-R kinase. Recent X-ray studies of closely related structures complexed with other kinases indicated that both binding modes could exist. This example showed that one must be prepared to consider more than one binding mode. This diversity created some complications in the analyses but also opened up a broader range of possibilities for the design of new structures.

ADDITIONAL CASE STUDIES¶
Additional Case Studies¶

Copyright © 2024 drugdesign.org