Case Studies in ADME/Tox Predictions¶
Info
Practical examples are presented in this chapter illustrating the exploitation of ADME/Tox data in drug discovery.
Number of Pages: 69 (±1 hours read)
Last Modified: June 2010
Prerequisites: None
ADME/Tox Case Study 1: Identification of Non-Genotoxic Carcinogens¶
Identification of Non-Genotoxic Carcinogens¶
The identification of non-genotoxic carcinogens is an important issue in ADME/Tox analyses; large species differences may occur for sub-sets of compounds. A model addressing this particular issue for both pharmaceutical and chemical compounds was introduced by Walter Westerink and Willem Schoonen at Schering-Plough. This is presented in the following pages.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Current ADME/Tox Analyses¶
Currently, carcinogens and non-genotoxic carcinogens are identified by means of in vivo and in vitro analyses.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Possible Models for Non-Genotoxic Carcinogens¶
Two classes of non-genotoxic carcinogens are relevant for this kind of toxicity. These carcinogens can bind to two specific classes of nuclear receptors, which are well known as: Arylhydrocarbon Receptor (AhR), and Peroxysomal Proliferator Activating Receptor alpha (PPARα).

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Both Receptors Form Heterodimers¶
The Arylhydrocarbon Receptor forms heterodimers with the Arylhydrocarbon Receptor Nuclear Translocation (ARNT) protein, whereas the Peroxysomal Proliferator Activating Receptor alpha forms a heterodimer with the Retinoic acid X Receptor (RXR). Three different RXR forms exist, known as RXRα, RXRβ, and RXRγ. Interaction with all three forms is possible, but RXRα is most commonly described in this aspect.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Activation for the Arylhydrocarbon Receptor¶
The mechanisms of activation for the Arylhydrocarbon Receptor (AhR) is as follows: A ligand enters the cell by diffusion and binds to AhR after dissociation of the Heat Shock Protein 90 (HSP90). This AhR occupied with ligand will pass through the nuclear membrane and forms a heterodimer with the AhR Nuclear Translocation (ARNT) protein. This heterodimer can finally bind to the specific Responsive Element (RE) in the promoter of the target gene and transcribe and translate many enzymes, such as cytochrome P450 variants CYP1A1, CYP1A2 and CYP1B1.
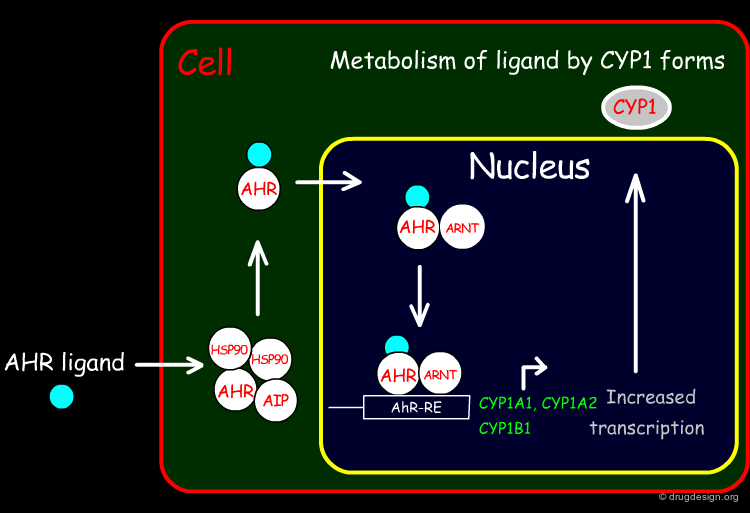
Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
The Responsive Elements of Interaction (1/2)¶
The responsive elements of AhR and PPARα interaction as well as of estrogen (E), glucocorticoid (GT), thyroid hormone (T) and retinoic acid (RA) receptors are known. For interaction of nuclear receptors specific interaction sites exist in the promoter of specific genes. For homo- and heterodimers these sequences exist of 5 to 6 base pairs in an inversed repeat or mirror image. Characteristic examples are shown in the figure below and are explained in the following page.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
The Responsive Elements of Interaction (2/2)¶
These 5 to 6 base pairs (in red) are very important for the interaction of the first zinc finger of the nuclear receptor with the DNA. A small change of one of these central base pairs 3 and/or 4 can change the activity from being active for estrogens in becoming active for glucocorticoids. Moreover, the space between the responsive elements is also very important, as shown for ERE and TRE, which contain either 3 different base pairs or none at all. In case of the AhR-RE the outside flanking of the 5 base pairs GCGTG is also very relevant for the recognition of AhR.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Binding Analysis of the Arylhydrocarbon Receptor (AhR)¶
The most common ligand is identified as dioxin. The chemical catastrophe in Seveso in Italy in the seventies resulted in a large exposure of dioxin. Due to this exposure many people, who came in contact with dioxin, suffered years later from the increases of tumor incidence. When the AhR was identified, assays were developed to measure the exposure towards dioxin. Binding studies with dioxin (TCDD) and 3-methylcholanthrene (3MC) are one way to identify this AhR (Riddick et al. 1994). The chemical structures of these ligands are shown on the next page.
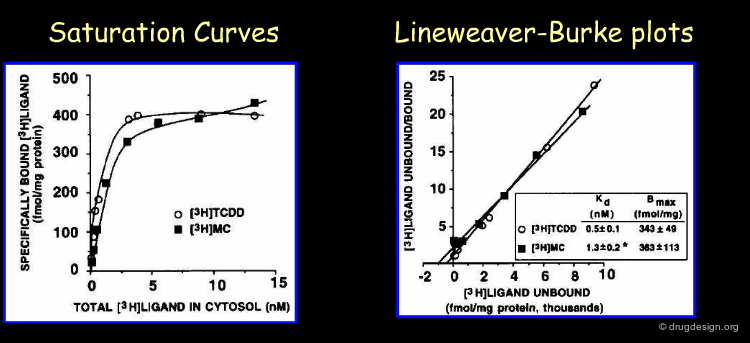
Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
articles
2,3,7,8-Tetrachlorodibenzo-p-dioxin versus 3-methylcholanthrene: comparative studies of Ah receptor binding, transformation, and induction of CYP1A1 Riddick DS, Huang Y, Harper PA, Okey AB J Biol Chem 269(16) 1994
Ligand Identification of the Arylhydrocarbon Receptor (AhR)¶
Besides dioxin, also known under the name 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), more ligands were identified for the AhR. The ligands indicated in the figure below are also very particular. β-Napthoflavone is present in vegetables, while Benzo[a]pyrene is present in the smoke of a cigarette and burned meat. Polychlorobivinyls are chemical waste products, while Induribin is a trypthophan derivative and Indigo a blue color dye.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Induction on the mRNA Level by AhR¶
The mRNA induction levels of CYP1A1 and CYP1A2 are observed and expressed as fold change over the control. The measurements are performed by Q-PCR for several AhR agonists at different concentration levels in human liver hepatocytes in HepG2 cells (Westerink and Schoonen, 2007). If we look at the lowest concentration dependent inductions (Blue), we clearly see that TCDD is the most potent inducer for both CYP1A1 and CYP1A2, followed by 3-MC. Indirubin and Indigo, on the other hand, became active at 10-8 M, and BNF at 10-7 M.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
articles
Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells Westerink WM, Schoonen WG. Toxicol In Vitro 21(8) 2007
Induction on the Enzyme Level by AhR¶
Also, the enzyme induction levels of CYP1A1 and CYP1A2 are observed and expressed as luminescence change. The measurements are performed with specific Promega substrates for CYP1A1 and CYP1A2 in combination with several AhR agonists at different concentration levels in human liver hepatocytes in HepG2 cells (Westerink and Schoonen, 2007). Again if we look at the lowest concentration dependent inductions (Blue), we clearly see that TCDD is the most potent inducer for both CYP1A1 and CYP1A2, followed by 3-MC. Indirubin, Indigo and BNF, on the other hand, became active at 10-8 M.
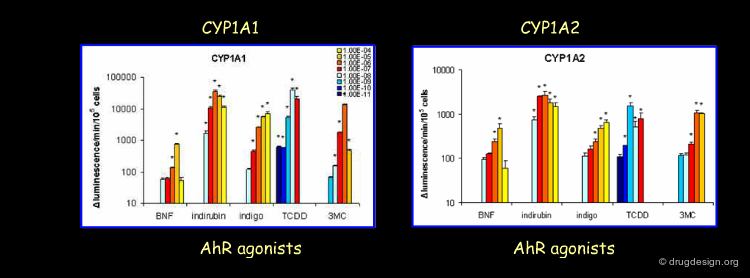
Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
articles
Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells Westerink WM, Schoonen WG. Toxicol In Vitro 21(8) 2007
Similar Induction of Enzyme Activity Between Species (1/2)¶
The induction of the enzyme activities of CYP1A1 and CYP1A2 can be relatively similar between different species (Westerink et al., 2008). This is the case for many AhR agonists, as shown below. For human HepG2 and rat H4IIE hepatocytes. TCDD is the most potent inducer in both the human and rat cell line. TCDD is equipotent for both cell lines as 3-MC is.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
articles
Pharmacologic profiling of human and rat cytochrome P450 1A1 and 1A2 induction and competition Westerink WM, Stevenson JC, Schoonen WG. Arch Toxicol 82(12) 2008
Similar Induction of Enzyme Activity Between Species (2/2)¶
Differences in strength can easily occur. PCB126 is a 1000-fold and indigo a 100-fold more active inducer for the rat hepatocyte, while Indirubin is a 100-fold more potent inducer for the human hepatocyte. Moreover, also benzo[a]pyrene and BNF were weaker inducers, both were still 100-fold more potent in rat than in human hepatocytes. At higher concentrations the compounds may become toxic, as indicated by the lower response rates.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
articles
Pharmacologic profiling of human and rat cytochrome P450 1A1 and 1A2 induction and competition Westerink WM, Stevenson JC, Schoonen WG. Arch Toxicol 82(12) 2008
Examples of Rat Specific Induction of Enzyme Activities¶
Rat specific induction of the enzyme activities of CYP1A1 and CYP1A2 be completely different from other species. One of these compounds Org D appeared very carcinogenic in the rat in vivo setting. However, these compounds may appear to be very rat specific. These compounds will be called non-genotoxic carcinogens in rats, but may be irrelevant for the human setting. Rat specific carcinogenic studies will result in the wrong answers for humans.
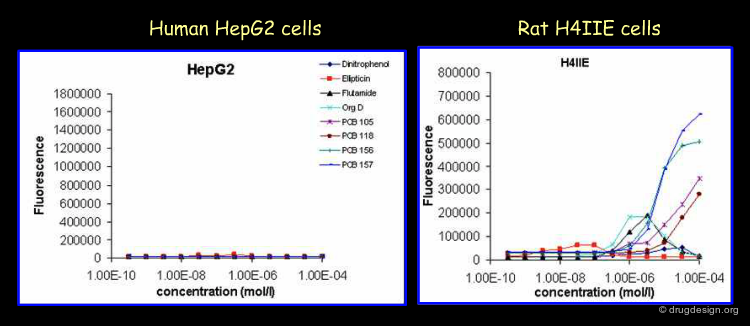
Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Examples of Human Specific Induction of Enzyme Activities¶
Human specific induction of the enzyme activities of CYP1A1 and CYP1A2 be completely different from other species. This is the case for some AhR agonists. These compounds will be negative negative in in vivo carcinogenicity studies in rats, but may be human specific carcinogens. Rat specific in vivo carcinogenicity studies will result in the wrong answers for humans.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Conclusion¶
In vitro analysis, but not in vivo analysis for carcinogenesis in rat only, may result in the proper decision for non-genotoxic carcinogenic compounds. Therefore the identification of non-genotoxic carcinogens in humans can best be carried out with the following procedures, as summarized below.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
Outcome of this Testing¶
The outcome of this testing is determinant for deciding further development of the drug candidate under consideration.

Author
Willem Schoonen and Walter Westerink Department of Pharmacology, DMPK and Safety, Schering-Plough, Oss, The Netherlands
ADME/Tox Case Study 2: Interpretation of Toxicology from an ADMET Standpoint¶
Case Studies Presented Here¶
The case studies presented in the following pages aim at illustrating why a given drug can show marked species differences because of differences in pharmacological and pharmacokinetic (ADME) properties.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
The Action of a Drug¶
To understand that, one has to keep in mind that the global action of a drug results of the combination of both pharmacokinetic (ADME) and pharmacodynamic properties.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Reasons for Species Specific Responses¶
Related drugs may have entirely different species specific responses for reasons that are listed in the figure below. In the following pages examples are given to illustrate some of them.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
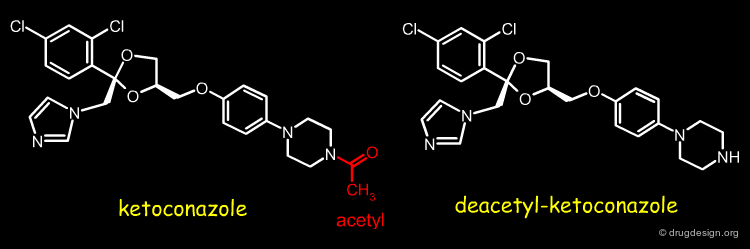
Example-1: Differences in Metabolism¶
For two analog drugs the metabolic products are in general the same. In some cases different metabolic products are formed resulting in differences in species specific responses. This is the case of ketoconazole and its deacetyl-decotonazole analog.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Different Metabolism of the two Analogs¶
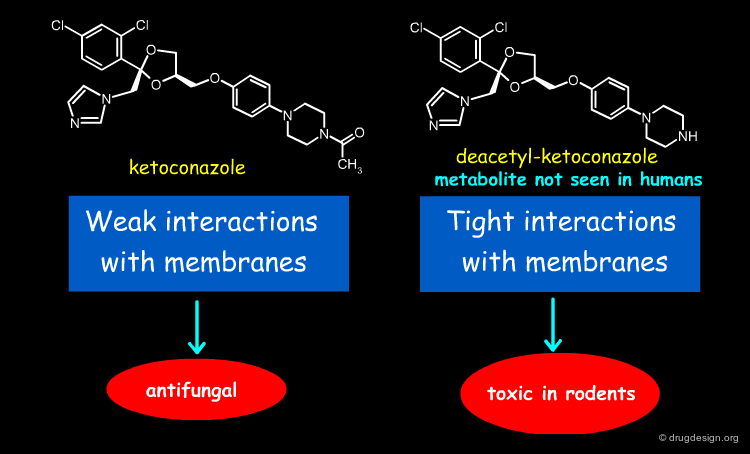
Ketoconazole is a lipophilic antifungal drug rendered weakly basic by the presence of an immidazole grouping. Metabolism in humans is mainly by N-glucuronidation of this function rendering the compound much more water soluble. Rodents have very high amidase and esterase levels and the principal metabolites formed are by hydrolysis of the acetyl moiety. The resultant secondary amine renders this metabolite much more basic (and now ionised substantially at physiological pH than the parent ketoconazole, but still with substantial lipophilicity. The combination of lipophilicity and basicity is associated with substantial membrane interactions to the extent that toxicity in the form of phospholipidosis is observed.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Ketoconazole Binding¶
In the phospholipid bilayer, ketoconazole orientates itself in order to maximize the hydrophobic interactions between its dichlorophenyl group and its acylated piperazine together with the long alkyl side chains of the lipid layer of the membranes (see figure below). This corresponds to weak binding interactions.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Mode of insertion of miconazole, ketoconazole and deacylated ketoconazole in lipid layers. A conformational analysis Brasseur, R., C. Vandenbosch, H. Van den Bossche, and J. M. Ruysschaert. Biochemical Pharmacology 32 1983
Desacetyl-Ketoconazole Binding¶
In desacetyl-ketoconazole the situation is different because of the existence of a positive charge on the piperazine ring. The molecule adopts a conformation reverted as compared to ketoconazole where the dichlorophenyl group is inserted in the lipid layer and the piperazine moiety is pointing towards the aqueous phase. In this case there are strong binding interactions.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Mode of insertion of miconazole, ketoconazole and deacylated ketoconazole in lipid layers. A conformational analysis Brasseur, R., C. Vandenbosch, H. Van den Bossche, and J. M. Ruysschaert. Biochemical Pharmacology 32 1983
Example-2: Differences in Rate of Metabolism¶
Differences in rate of metabolism may lead to different dose response relationships. This is a common finding when comparing animals and human. Proxicomil is one such example, the compound is slowly metabolized in dogs and rapidly metabolized in rats and humans.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Metabolites in Safety Testing (MIST): Considerations of Mechanisms of Toxicity with Dose, Abundance, and Duration of Treatment. Smith D.A. and Obach R.S. Chem. Res. Toxicol. 22 2009
PK /PD Model of Response¶
Due to the slow rate of metabolism concentrations of proxicromil are much higher for an equivalent dose in the dog than in rats and human. This results in exaggerated pharmacological and toxicological effects for equivalent doses in the two species.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Origin of the Hepatotoxicity of Procicromil in Dog¶
The differences in metabolism with Proxicromil result in it being hepatotoxic in dog. Further understanding is provided by considering the structures of proxicromil and its metabolite. The rapid hydroxylation in the liver in rat and man results in lowering the lipophilicity of the molecule and removing its amphipathis nature (detoxification). The hydroxylation is very slow in dog resulting in high concentrations of the detergent like proxicromil compound at the biliary canaliculus within the liver.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Chromone-2-Carboxylic Acids: Roles of Acidity and Lipophilicity in Drug Disposition Smith, D.A., Brown, K., Neale, M.G. Drug Met.Rev. 16 1985-1986
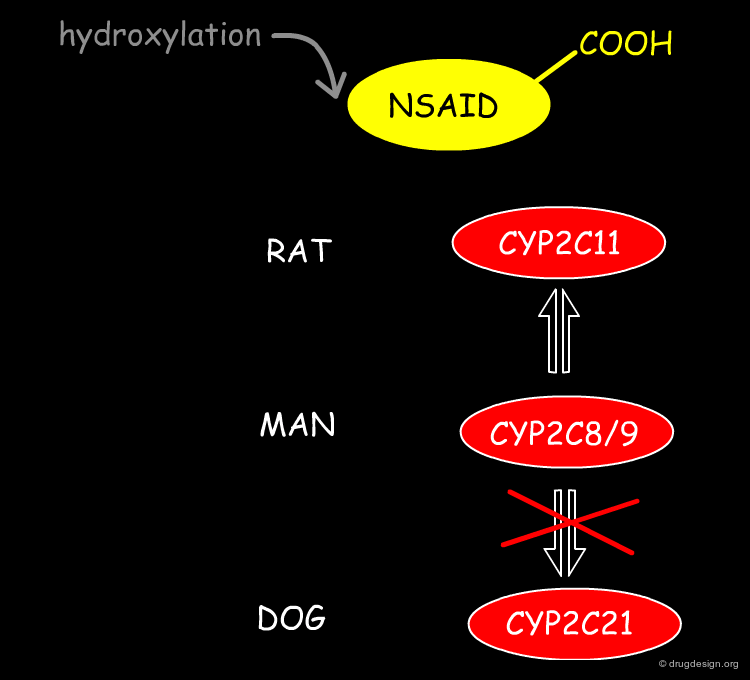
Toxicity due to Different Plasma Clearance Values¶
The enzymes that catalyse the oxidation of proxicromil are the CYP2C family with the dog homologue having a much lower rate of metabolism. Agents such as tolbutamide, diclofenac, tienilic acid, torasemide, bumetamide, probenicid, fenoprofen, flurbiprofen, etodolac, sultosilic acid, bucloxic acid and proxicromil are acidic and are CYP2C substrates. They are all slowly hydroxylated in dogs as compared to rat and humans. This results in potentially exaggerated pharmacological and toxicological effects at equivalent doses.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Integration of animal pharmacokinetic and pharmacodynamic data in drug safety assessment Smith D.A. Eur. J. Drug Met. and Pharmacokinet. 18 1993
Example-3: Differences in Receptor Affinity¶
Differences in receptors between different species are fairly common, although in many cases these diffences are not sufficiently large to not give broadly equivalent responses to a particular compound. For some receptors the differences are meked and dwarf the effects previously described for clearance and metabolism. A classical case is the differences of affinity for cardiac glycosides and the human and rat cardiac Na+/K+ ATPase. This is presented in the next page.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Pharmacokinetics and pharmacodynamics in toxicology Smith D.A. Xenobiotica 27 (5) 1997
Affinity for Cardiac Na+/K+ ATPase (1/2)¶
The following slide illustrates why the pharmacodynamics of cardiac glycosides in terms of cardio-pharmacology and cardio-toxicity in human are poorly predicted in rat. The comparison of affinity for cardiac Na+/K+ ATPase in human and rat is presented here where species differences in systemic clearance and affinity of cardiac glycosides (ouabain) for cardiac Na+/K+ ATPase are observed.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Affinity for Cardiac Na+/K+ ATPase (2/2)¶
Affinity is shown as reciprocal to give the figure a common axis. Over 4 log units span the differences in affinity (Human affinity being much higher, therefore concentrations in rat would need to be 10,000 times higher than those in man to elicit the same effect on the heart. Such high concentrations are probably not achievable and would also cause effects on other related proteins.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Interpretation of Toxicology from Animal Data¶
The interpretation of animal data and its extrapolation to man is complex and needs to be considered from pharmacokinetic and pharmacodynamic grounds. The simplest cases are where receptor profiles are similar in animals and man and adverse events in animals are separated by large multiples of dose and concentration from those to be used clinically. Even then progress in human is gradual and cautious.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Interpretation of Toxicology from Human Data¶
Even human is not a good model for human, as findings in as few as 1 in 30,000 patients, for a previously unseen toxicity, can lead to labelling warnings or even marketing withdrawal. Careful constant consideration of all the available data, from all viewpoints does though mitigate the risk.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
ADME/Tox Case Study 3: Drug Withdrawals due to Toxicity¶
Drug Failures, Lessons and Learnings for the Future¶
A survey of drugs withdrawn in past 25 years in the US market is presented in the following pages. The reasons for drug withdrawals are described and lessons that can be learned are considered.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Drug withdrawals and the lessons within DA Smith, EF Schmid Curr Opin Drug Discov Devel 9 (1) 2006
Editorial Review: Safety of drugs can never be absolute Smith DA, Johnson DE, and Park BK Curr Opin Drug Disc Dev 9(1) 2006
Adverse drug event surveillance and drug withdrawals in the United States, 1969-2002. The importance of reporting suspected reactions Wysowski DK and Swartz L Arch Intern Med 165 2005
Communicating risk/benefit of medicines in todays environment Schmid EF, Smith DA and Ryder SW Drug Discovery Today 12 2007
Three Types of Drug Withdrawals¶
The 1980-2005 drug withdrawals can be grouped into 3 categories. The drug toxicities were analyzed and classified based on the FDA AERS system (Adverse Event Reporting System) which consists of a primary surveillance database for post-marketing drug safety issues. They are shown in the table below; the fourth type of withdrawal (type D) is not considered here because there are no examples of drugs withdrawn for Type D toxicity over the period covered by this review.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Adverse drug event surveillance and drug withdrawals in the United States, 1969-2002: The importance of reporting suspected reactions Wysowski DK, Swartz L Arch Intern Med 165(12) 2005
Type A¶
Drug withdrawal of Type A: the toxic mechanism has a pharmacological basis. There are two subtypes of type A; one based on the primary pharmacology (type A1) of the drug (i.e. selective activity against the intended target of the drug) and the other based on other secondary pharmacology(s) (type A2) of the drug (non-selective activity against non-intended receptors, enzymes, ion channels etc.).

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type B¶
Drug withdrawal of Type B: the toxicity probably has an immunological base. Such toxicities do not necessarily exhibit classic dose-response relationships. They are generally observed in a very low proportion of patients and are suspected to involve a reaction between drug or metabolite and tissue macromolecules.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type C¶
Drug withdrawal of Type C toxicity, is similar but the effect of the drug is also due to a chemical reaction between drug or metabolite and tissue macromolecules but there is a rapidly ensuing response. Similarly this toxicity follows a more classical dose response relationship.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type D¶
Drug withdrawal of Type D refers to toxicity in which duration or time of administration is critical and the response is delayed, even for years. Examples of type D toxicity include carcinogenesis and teratogenesis. As indicated above, there are no examples of drugs withdrawn for Type D toxicity over the last 25 years; we are therefore mainly concerned in this review with types A, B and C.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Table of Drug Withdrawals from 1980¶
Drug withdrawals from 1980 to date and reason for withdrawal are indicated in the table below.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type A1 Drug Withdrawals¶
In the following pages are presented drugs withdrawn for problems on the Type A1 primary pharmacology of the drug.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
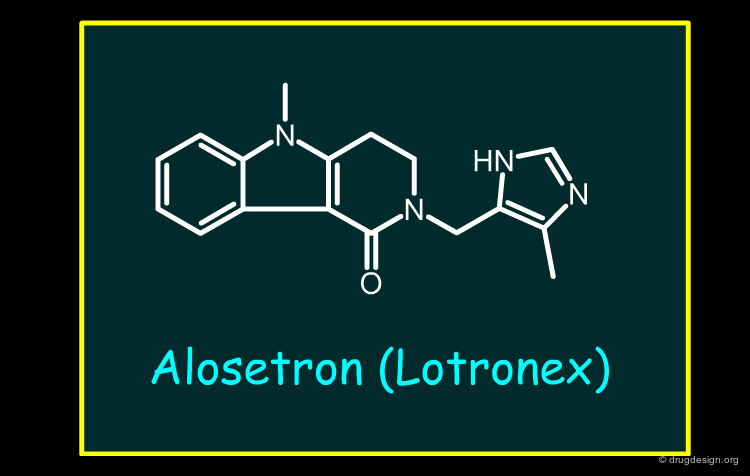
Alosetron¶
Alosetron is a 5-HT3 antagonist introduced for the treatment of irritable bowel syndrome (IBS). 5-HT3 antagonists suppress the reflex activation of colonic motor function in response to food ingestion and can cause constipation as a side effect. The withdrawal of the drug was due to several cases of ischemic colitis. Constipation can be one of the factors that facilitate the development of ischemic colitis. Alosetron was subsequently re-introduced in 2002, following a risk/benefit assessment, but is now significantly restricted in use.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Pharmacology and clinical experience with alosetron Camilleri M Exp Opin Invest Drugs 9(1) 2000
Possible mechanisms for ischemic colitis during alosetron therapy Beck IT Gastroenterology 121(1) 2001
Cerivastatin¶
Cerivastatin was the most potent of a group of drugs inhibiting liver hydroxymethyl glutanyl-coenzyme A (HMG-CoA) termed statins. Muscle toxicity (myopathy) is a low incidence adverse effect of statins, the extreme form of which is rhabdomyolysis which can lead to kidney failure and death. Cerivastatin showed rates of rhabdomyolysis up to 10 fold greater than other HMG-CoA inhibitors. The systemic bioavailability was the highest of any statin. Bioavailability in this context could be viewed as a ratio of systemic burden (muscle-toxicity) versus hepatic burden (liver-efficacy) and may point to a key factor that differentiates this drug.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Rhabdomyolysis associated with hydroxymethylglutanyl-coenzyme A reductase inhibitors Jamal SM, Eisenberg MJ, Christopoulos S Am Heart J 147(6) 2004
Flosequinan¶
Flosequinan is a vasodilator with mixed arterial and venous effect. The agent had mild positive chronotropic and ionotropic effects. The drug has an active sulphone metabolite, Flosequinoxan, which also contributes to the effect and may be the active principle. Some of the pharmacology of Flosequinan and its metabolite may relate to its activity as a PDE III inhibitor. Flosequinan was associated with an excess mortality rate. It is probable that the detrimental effects of flosequinan are multifactorial but the drug increased circulating noradrenaline concentrations and heart rate and these may have been contributing factors in the mortality observed.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Neurohormonal activation in severe heart failure: Relations to patient death and the effect of treatment with flosequinan Moe GW, Rouleau JL, Charbonneau L, Proulx G, Arnold MO, Hall C, de Champlain J, Barr A, Sirois P, Packer M Am Heart J 139(4) 2000
Positive inotropic and hemodynamic properties of flosequinan, a new vasodilator, and a sulfone metabolite Falotico R, Haertlein BJ, Lakas-Weiss CS, Salata JJ, Tobia AJ J Cardiovasc Pharmacol 14(3) 1989
Studies on the cardiac actions of flosequinan in vitro Gristwood RW, Beleta J, Bou J, Cardelus I, Fernandez AG, Llenas J, Berga P Br J Pharmacol 105(4) 1992
Pharmacokinetics of flosequinan in patients with heart failure Nicholls DP, Droogan A, Carson CA, Taylor IC, Passmore AP, Johnston GD, Kendall M, Dutka D, Morris GK, Underwood LM, Hind ID Eur J Clin Pharmacokinet 50(4) 1996
Encainide¶
Encainide was an anti-arrhythmic agent which blocked the Na+ channel excitatory current. A major series of studies (CAST) examined whether drugs of this type could prevent sudden death in high-risk patients. These patients had frequent premature ventricular extrasystoles (PVCs) and reduced ventricular function following acute myocardial infarction. The placebo group was found to have lower mortality compared to the group treated with Encainide (and Flecainide). The excess number of new ischemic events in the placebo group equaled the excess number of deaths in the treated group indicating that new ischemia was converted to sudden death.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
From first class to third class: Recent upheaval in antiarrythmic therapy - lessons from clinical trials Lazzara R Am J Cardiol 78(4A) 1996
Rofecoxib¶
Rofecoxib (Vioxx) belongs to the anti-inflammatory class of drugs termed COX-2 inhibitors. These were compounds with advantages over conventional non-selective COX inhibitors (NSAIDs) since the gastrointestinal effects such as ulceration, and internal bleeds are mediated by COX-1. Rofecoxib increased the risk of thrombotic cardiovascular events. A pharmacological rationalisation of this effect can be advanced concerning suppression of endothelial vascular PGI2 whilst not inhibiting platelet derived TXA2 production resulting in an imbalance of homeostatic, normally endothelium protective and antithrombotic, mechanisms.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Development of COX-2 selective inhibitors - therapeutic perspectives Casturi SR, Hegde P, Ramanujam R Curr Med Chem - Immunol Endocrin Metab Agents 5(3 2005
Lessons from Withdrawals due to Primary Pharmacology¶
It is extremely difficult to draw simple conclusions from this class of market withdrawal. The obvious reaction of Regulatory Authorities and Pharmaceutical Companies is to study the drug more extensively in Development: "outcomes" trials now form part of Development rather than a post-marketing activity, contributing hugely to the cost of drug development.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
A Broad Spectrum of Drugs¶
The period covered (1980 to date) spans "phenomena" type drugs, such as Flosequinan to highly specific selective drugs such as Alosetron. Phenomena drugs do not have a specific target or mechanism. Screening of compounds is normally by pharmacological effect and the drugs therefore may have multiple mechanisms. Although it may be possible to separate the beneficial and deleterious effects, scientifically this is very difficult. This invariably makes the design of newer agents with improved therapeutic ratios almost impossible. Alosetron represents the other extreme where the selectivity is almost absolute and the side effects must be associated with the primary pharmacology.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Impact on Decisions in Drug Discovery¶
It is extremely unlikely that a company would progress a follow-on drug on the off chance that it may be better.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type A2 Drug Withdrawals¶
In the following pages are presented drugs withdrawn for problems on the Type A2 primary pharmacology of the drug.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
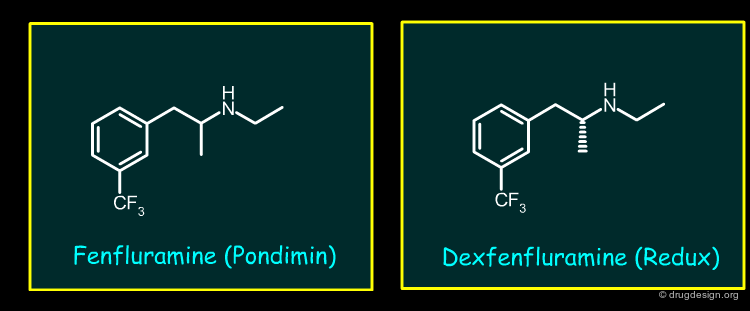
Fenfluramine and Dexfenfluramine (1/2)¶
The appetite suppressants Fenfluramine and Dexfenfluramine (single enantiomer of Fenfluramine), both effective in the treatment of obesity, were withdrawn from the marketplace because of association with pulmonary hypertension and valvular heart disease. Fenfluramine and Dexfenfuramine bind weakly to 5-HT receptors.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Possible role of valvular serotonin 5-HT2B receptors in the cardiopathy associated with fenfluramine Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL et al Mol Pharmacol 57(1) 2000
Evidence for possible involvement of 5-HT2B receptors in cardiac valvulopathy associated with fenfluramine and other serotonergic medications Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL Circulation 102(23) 2000
Fenfluramine and Dexfenfluramine (2/2)¶
A major metabolite Norfenfluramine is responsible for the pharmacological activity. The drugs reduce appetite by activating 5-HT2C receptors. Norfenfuramine binds and potently activates 5-HT2C receptors but is equally potent as an agonist of 5-HT2B receptors. Activation of mitogenic 5-HT2B receptors on heart valves and pulmonary artery interstitial cells lead to the formation of proliferative fibromyxoid plaques that cause the deleterious changes in tissue integrity and function.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Rapacuronium¶
Rapacuronium was introduced as an ultra-short acting nondepolarising neuromuscular blocking agent of nicotinic receptors. The drug was used to facilitate intubation. The drug was withdrawn from clinical use due to a high incidence of bronchospasm, sometimes with lethal consequences. Rapacuronium is an antagonist of M2 muscarinic receptors. Normally the release of acetylcholine is terminated by the feedback of M2 receptors. Selective antagonism of these receptors leads to excessive release of acetylcholine, which can then act on the M3 receptors in airway smooth muscle, causing bronchospasm.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
A mechanism for rapacuronium-induced bronchospasm Jooste E, Klafter F, Hirshman CA, Emala CW Anesthiology 98(4) 2003
Rapacuronium preferentially antagonizes the function of M2 versus M3 muscarinic receptors in guinea pig airway smooth muscle Jooste E, Zhang Y, Emala CW Anesthiology 102(1) 2005
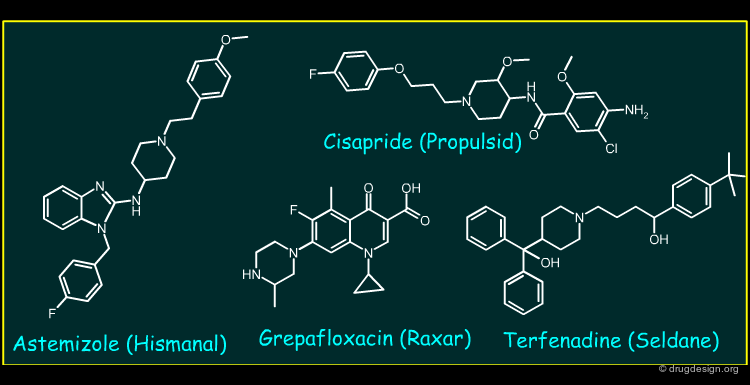
Astemizole, Cisapride, Grepafloxacin and Terfenadine (1/2)¶
Astemizole, Cisapride, Grepafloxacin and Terfenadine are drugs that represent a pharmacologically diverse group. The primary pharmacology of Astermizole and Terfenadine are non-sedating H1 anti-histamines used to treat various allergic conditions. Cisapride is a gastro-intestinal pro-kinetic motility agent acting by activating 5-HT4 receptors, whilst Grepafloxacin is a fluoroquinolone antibiotic.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Astemizole, Cisapride, Grepafloxacin and Terfenadine (2/2)¶
All the compounds inhibit the rapidly activating delayed rectifier potassium current (channel) (IKr) in cardiac tissues. The slowing of repolarisation leads to QT interval prolongation and the appearance of potentially fatal arrythmias (torsade de pointes, "TdP").

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Microarray analysis in human hepatocytes suggests a mechanism for hepatotoxicity induced by trovafloxacin Liguori MJ, Anderson LM, Bukofzer S, McKim J, Pregenzer JF, Retief J, Spear BB, Waring JF Hepatology 41(1) 2005
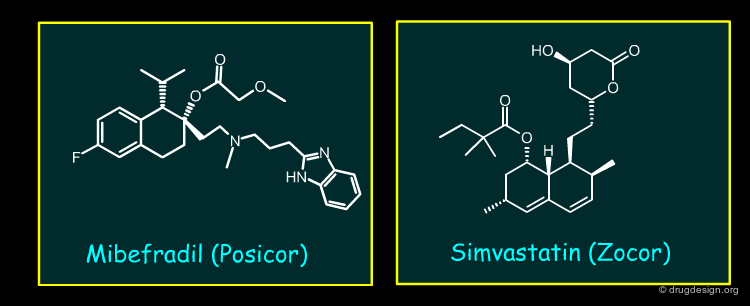
Mibefradil¶
Mibefradil is a long acting T-type calcium channel antagonist used for hypertension and chronic stable angina pectoris. Mibefradil was a potent inhibitor of CYP3A4, which resulted in multiple clinically relevant drug interactions. These included very important co-medications such as Simvastatin, which showed a large seven-fold elevation, sufficient to enhance potential adverse events, including rhabdomyolysis.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Clinical pharmacokinetics of mibefradil Welker HA, Wiltshire H, Bullingham R Clin Pharmacokinet 35(6) 1998
Metabolic interactions between mibefradil and HMG-CoA reductase inhibitors: An in vitro investigation with human liver preparations Prueksaritanont T, Ma B, Tang C, Meng Y, Assang C, Lu P, Reider PJ, Lin JH, Baillie TA Br J Clin Pharm 47(3) 1999
Lessons from Withdrawals Due to Secondary Pharmacology¶
The impact of lack of specificity of a drug is obvious with hindsight and can be rationalised and incorporated into screening cascades. This is particularly appropriate today, since almost all drug discovery programmes are mounted using the "pure" protein as the primary screen. Close neighbours to the desired target as highlighted with Fenfluramine/Desfenfluramine, will be screened alongside the primary pharmacology.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Impact on Discovery Screening Programs¶
Other important screens such as IKr will also be placed high in the cascade particularly when the structure and physiochemistry of the lead chemical series makes interactions with this channel likely. Screens can be configured in high throughput format. Similarly screening for CYP inhibition takes place high in the cascade. Initially most screens examined reversible inhibition, but mechanism based inhibition, of the type seen with mibefradil, is now routinely incorporated. Broad ligand binding is also widely adopted at a later stage in drug discovery.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type B and C Toxicity Drug Withdrawals (1/4)¶
Benoxaprofen, Bromfenac, Ticrynafen, Troglitazone, Tolcapone and Trovafloxacin were withdrawn from the market place due to hepatotoxicity.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Type B and C Toxicity Drug Withdrawals (2/4)¶
Temafloxacin, Nomifensine and Remoxipride were withdrawn due to haemolytic and aplastic anaemia.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Caspase-dependent and -independent mechanisms in apoptosis induced by hydroquinone and catechol metabolites of remoxipride in HL-60 cells Inayat-Hussain SH, McGuinness SM, Johansson R, Lundstrom J, Ross D Chem-Biol Interact 128(1) 2000
Characterization of glutathione conjugates of the remoxipride hydroquinone metabolite NCQ-344 formed in vitro and detection following oxidation by human neutrophils Erve JC, Svensson MA, von Euler-Chelpin H, Klasson-Wehler E Chem Res Toxicol 17(4) 2004
High sensitivity determination of the remoxipride hydroquinone metabolite NCQ-344 in plasma by coupled column reversed-phase liquid chromatography and electrochemical detection Nilsson LB Biomed Chromatogr 12(2) 1998
Induction of apoptosis by remoxipride metabolites in HL60 and CD34+/CD19-human bone marrow progenitor cells: Potential relevance to remoxipride-induced aplastic anemia McGuinness SM, Johansson R, Lundstrom J, Ross D Chem-Biol Interact 121(3) 1999
Type B and C Toxicity Drug Withdrawals (3/4)¶
Suprofen was withdrawn due to flank pain syndrome and Zomepirac due to anaphylaxis. The flank pain syndrome is accompanied by renal dysfunction and associated changes in the excretion of uric acid. In many of the cases more than one toxicity finding also contributed to the withdrawal. For instance Temafloxacin was also associated with renal and liver toxicity and Ticrynafen also exhibited flank pain syndrome.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Mechanism-based inactivation of human recombinant P450 2C9 by the nonsteroidal anti-inflammatory drug suprofen O'Donnell JP, Dalvie DK, Kalgutkar AS, Obach RS Drug Metab Dispos 31(11) 2003
Suprofen-induced uricosuria. A potential mechanism for acute nephropathy and flank pain Abraham PA, Halstenson CE, Opsahl JA, Matzke GR, Keane WF Am J Nephrol 8(2) 1988
Type B and C Toxicity Drug Withdrawals (4/4)¶
Furthermore, some of these drugs such as Nomifensine or Troglitazone were withdrawn from a market that had safer, effective follow-on medicines available.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
# Lessons Learned from Type B/C Toxicity¶
In many cases of direct organ toxicity reactive metabolites, which co-valently bind to macromolecules are implicated in the mechanism. Certain groups or functions in molecules are particularly susceptible to metabolism to reactive intermediates. These groupings are termed "structural alerts". Of particular note are aromatic amines (anilines) and thiophenes. Such groupings occur in Bromfenac, Nomifensine, Suprofen and Ticrynafen.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
A review of the common properties of drugs with idiosyncratic hepatotoxicity and the "multiple determinant hypothesis" for the manifestation of idiosyncratic drug toxicity Li AP Chem-Biol Interact 142(1-2) 2002
A comprehensive listing of bioactivation pathways of organic functional groups Kalgutkar AS, Gardner I, Obach RS, Shaffer CL, Callegari E, Henne KR, Mutlib AE, Dalvie DK, Lee JS, Nakai Y, O'Donnell JP et al Curr Drug Metab 6(3) 2005
Structure toxicity relationships - how useful are they in predicting toxicities of new drugs? Nelson SD Adv Exp Med Biol 500 2001
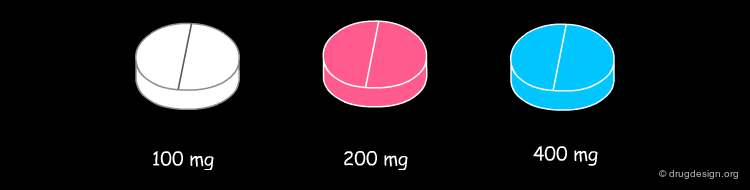
Importance of Dose in B and C Toxicity¶
Another feature of direct organ toxicity is that the causal drugs are often administered at a high dose usually of 100 mg or greater. It is noticeable that all the drugs are given at 100 mg per day or greater and that 6/11 are administered at doses above 400 mg per day. Optimisation of compounds in drug discovery can aim at low doses (optimal pharmacokinetics, high intrinsic potency) without obvious structural alerts will attenuate the risk.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
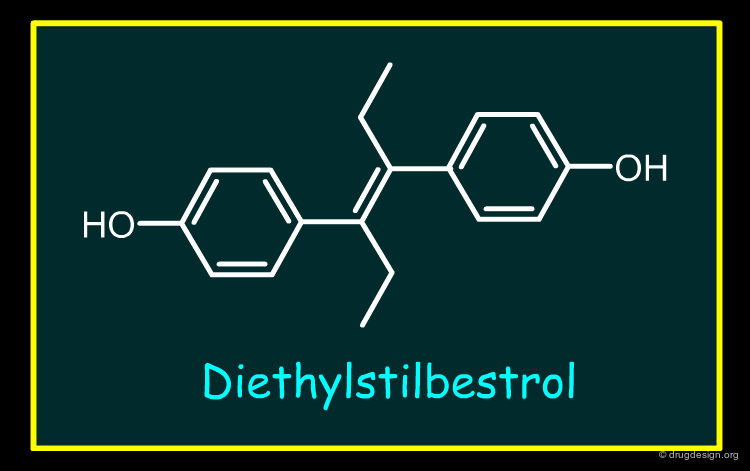
Type D Toxicity Drug Withdrawals¶
There are no examples of Type D withdrawals over the last 25 years. There are very few examples of drugs withdrawn due to human carcinogenicity. The only well documented is Diethylstilbestrol (withdrawn prior to the period covered by this review). With this drug primary pharmacology (A1) plays a major, although not necessarily the only role. Several factors lead to this conclusion: (I) studies in animals (mice) are able to recreate the changes in reproductive tract structure and function including malignant abnormalities seen in human; (II) these changes are dose related in terms of severity and (III) the changes are caused not just by Diethylstilbestrol but other estrogenic compounds (e.g. environmental estrogenic pollutants).
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Lessons learned from perinatal exposure to diethylstilbestrol Newbold RR Toxicol Appl Pharmacol 199(2) 2004
Thalidomide¶
An example of a drug withdrawn because of teratogenesis is Thalidomide (withdrawn prior to the period covered by this review) and the mechanism is still not fully defined. During the 25 year period a number of drugs received marketing authorisations which were teratogenic in animals. Such drugs receive labelling outlining the risk of administration to females.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
A novel hypothesis for thalidomide-induced limb teratogenesis: Redox misregulation of the NF-kappa-B pathway Hansen JM, Harris C Antiox Redox Signal 6(1) 2004
Theoretical basis for the activity of thalidomide Meierhofer C, Dunzendorfer S, Wiedermann C BioDrugs 15(10) 2001
Balancing Benefit / Risk¶
No matter how extensively tested a drug is prior to marketing a full understanding of its benefit / risk can only be approached after perhaps 100,000 patients have received the drug for an extensive period. Even then it is often extremely difficult to balance benefit / risk since in some cases many patients may benefit, but a small subset of patients (perhaps as low as 1 in 30,000) may be harmed.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
articles
Drug withdrawals and the lessons within DA Smith, EF Schmid Curr Opin Drug Discov Devel 9 (1) 2006
Editorial Review: Safety of drugs can never be absolute Smith DA, Johnson DE, and Park BK Curr Opin Drug Disc Dev 9(1) 2006
Adverse drug event surveillance and drug withdrawals in the United States, 1969-2002. The importance of reporting suspected reactions Wysowski DK and Swartz L Arch Intern Med 165 2005
Communicating risk/benefit of medicines in todays environment Schmid EF, Smith DA and Ryder SW Drug Discovery Today 12 2007
Benefit Analyses for Antidepressants¶
Availability of similar drugs in a class is a great help, as benefit across the class is approximately constant allowing risk to be more objectively compared. In the example below all anti-depressants are associated with a very low but real risk of hepatotoxicity. Nefazodone risk is substantially greater than the other drugs.

Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Monitoring¶
In almost all cases awareness of the risks, by education of physicians and patients and therefore better monitoring would help alleviate the extreme cases, and as in the case of Alosetron allow re-introduction of the drug.
Author
Professor Dennis A. Smith Academic Research Fellow supported by Pfizer, former vice President of Pharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Sandwich, UK
Copyright © 2024 drugdesign.org