Case Studies in Advanced Analog Design¶
Info
This chapter presents examples illustrating intelligent attempts at analog design. It includes examples of scaffold morphing for the creation of pseudo-rings that mimic a real one and vice-versa, and the rational combination of molecular modeling and bioisosterism.
Number of Pages: 91 (±2 hours read)
Last Modified: December 2008
Prerequisites: None
Case Study-1 : Salicylanilides¶
Salicylanilides¶
The story of the design of a new type of EGF-R protein kinase inhibitors by Novartis is presented here. The quinazoline compound such as 1 (AstraZeneca) is a very potent EGF-R protein kinase inhibitor. Two possible hypotheses for the binding mode of this type of structure are discussed in the section "introduction to protein-ligand binding" however the work that is presented here can be understood independently of the exact mode of binding of this molecule. The structure of this molecule was modified by scaffold morphing, as explained in the following pages.

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
The 4-anilinoquinazoline class of inhibitors of the erbB family of receptor tyrosine kinases Denny WA Il Farmaco 56 2001
Tyrosine kinase inhibitors. 16. 6,5,6-tricyclic benzothieno[3, 2-d]pyrimidines and pyrimido[5,4-b-] and -[4,5-b]indoles as potent inhibitors of the epidermal growth factor receptor tyrosine kinase. Showalter HD, Bridges AJ, Zhou H, Sercel AD, McMichael A, Fry DW J. Med. Chem 42 1999
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of scytalone dehydratase Hodge CN and Pierce J Bioorganic and Medicinal Chemistry Letters 3 1993
Genistein Structure and Alignment with Quinazoline 1¶
The isoflavone genistein 2 is an unselective inhibitor of EGF-R protein kinase. The Novartis group observed that this structure superimposes well with that of the AstraZeneca quinazoline inhibitor 1. Moreover, the alignment reveals that a pseudo six-membered ring can be seen in the structure of genistein that mimics the pyrimidine ring of 1.


articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
3D Design of a Salicylanilide Scaffold¶
The analysis shown on the previous page prompted the Novartis team to consider a salicylanilide scaffold such as 3 (see button "2D") as a mimic of the quinazoline molecule 1. The formation of a strong hydrogen bond between the phenolic hydroxyl, and the carbonyl of the amide group in the salicylanilide structure maintains the molecule in the desired geometry.


articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Possible Intramolecular H-Bonds in Salicylanilides¶
Conformational analyses show that there are two possibilities of intramolecular H-bonds in the salicylanilide structure (3a and 3b). The high probability of the existence of the 3a conformation that mimics the molecular geometry of the AstraZeneca inhibitor 1 well, encouraged the Novartis team to consider the synthesis of several analogs of the salicylanilide structure.

articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Synthesis of the Molecules¶
About 30 analogs were synthesized; most of the analogs were prepared by reacting the corresponding salicyl acid with the intermediate formed from 3-chloroaniline with phosphorus trichloride. The synthesis used for the biphenylic anilide 28 is described below.
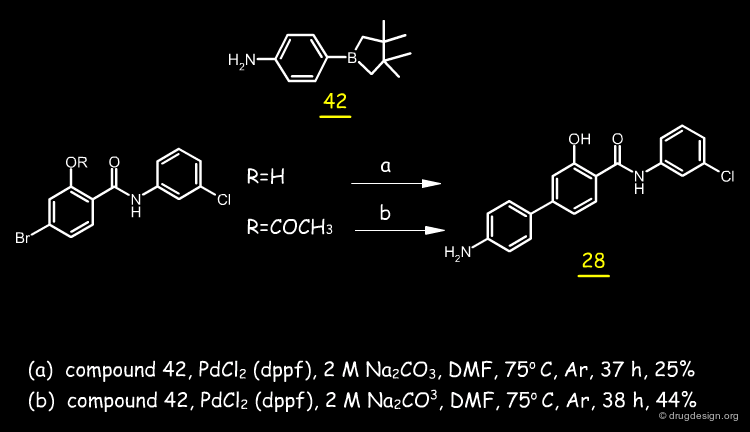
articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Biological Assays¶
The results obtained for certain compounds are shown below. Potent EGF-R kinase inhibitors that have an IC50 between 23 and 71 nM were found. Compounds that do not have a free phenolic hydroxyl group (such as 10 and 11) capable of making the intramolecular hydrogen bond with the carbonyl of the amide are not active.

articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Validity of the Hypotheses¶
The results confirmed the validity of the hypothesis that involved mimicking the AstraZeneca quinazoline by a salicylanilide structure. The two compounds have similar molecular geometries due to the existence of a pseudo-ring in the salicylanilide structure with a strong intramolecular hydrogen bond that maintains the proper geometry for the molecule.

articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Summary¶
The good superimposition in 3D of the structures of the AstraZeneca inhibitor 1 and genistein 2 revealed the mimicry of a pseudo-ring of genistein with the pyrimidine ring of 1. Following up on that observation, a salicylanilide scaffold 3 was imagined having the same molecular geometry as 1. This led to the discovery of a new class of potent EGF-R kinase inhibitors.

articles
Salicylanilides as inhibitors of the protein tyrosine kinase epidermal growth factor receptor Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P Eur. J. Med. Chem 39 2004
Case Study-2 : Pyrimidin-4-yl-ureas¶
Pyrimidin-4-yl-ureas¶
Novartis discovered a novel class of kinase inhibitors having a pyrimidin-4-yl urea chemotype. This structure was developed by P. Furet at Novartis through modeling. This molecule was designed by "scaffold morphing", a design process consisting of the progressive replacement of a real ring present in a reference lead compound by a pseudo ring. This is presented in the following pages.
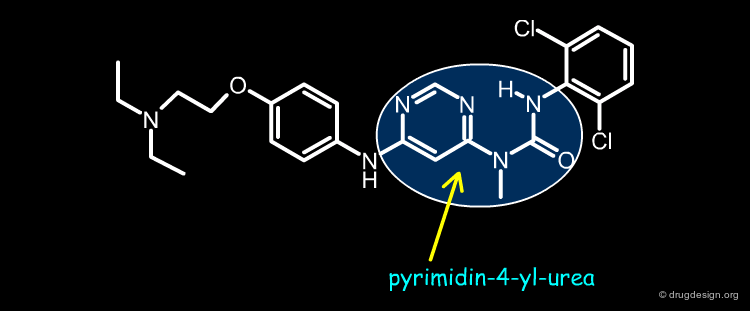
articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
PD-166285 Reference and Novartis Design¶
PD-166285 is a potent protein kinase inhibitor with a broad spectrum of activities and Novartis was interested in using this structure as a template to design new kinase scaffolds. The Novartis team realized they could move one nitrogen atom of the pyrazine heterocycle, and replace the pyrimidone ring by an urea moiety in order to form an intramolecular hydrogen bond which would maintain a global molecular geometry similar to that of the reference lead (see figure).

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
A Search in the Cambridge Structural Database¶
To test whether this type of molecular geometry can be found in small molecules, a search in the Cambridge Structural Database (CSD) using a pyrimidin-4-yl urea substructure was made. The search returned seven hits, that all had an intramolecular hydrogen bond between the pyrimidine ring and the urea.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Ab-Initio Calculations¶
Ab-initio calculations were made to assess the difference in energy between the extended and the folded pseudo-cyclic conformations of the model compound indicated below. The calculations in the gas phase showed that the pseudo-cyclic conformation was the most stable by 3.2 kcal/mol (populations at room temperature: 77% - 23%). When the calculations were made in water solution, the pseudo-cyclic form was favored by 0.5 kcal/mol (populations: 55% - 45%). This indicated that there would be no energy cost to pay for conformational adaptation to a kinase site.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Synthesis of the Prototype Molecule¶
The designed pyrimidyl urea 1 was prepared in two steps from commercially available materials.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Biological Assays for Pyrimidin-4-yl-urea¶
The pyrimidin-4-yl urea was tested in-vitro to assess its ability to inhibit the catalytic activity of several protein kinases. The activities observed are indicated below.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Docking of Pyrimidin-4-yl-urea in c-Abl¶
The pyrimidin-4-yl urea was docked in the ATP pocket of the c-Abl kinase. The 3D model revealed the existence of very tight interactions with the hydrophobic back pocket of the ATP binding site, in particular with the threonine gatekeeper residue.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Correlation of the Activities with Size of Gate Keeper¶
The existence of tight binding of the pyrimidin-4-yl urea with the c-Abl threonine gatekeeper prompted the Novartis team to analyze the nature of the gatekeeper residues in the different kinases. They found a good correlation between the biological activities and the volume of the gate keeper. Protein kinases having a gate keeper larger than threonine cannot be active.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Alignment of Pyrimidin-4-yl urea and PD-166285¶
Below is shown the good mimicry between the pyrimidin-4-yl urea structure 1 and PD-166285.


articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
P&G Discovered Independently the Same Molecule¶
It is interesting to note that the Procter and Gamble group independently discovered the same kinase inhibitor 1 (optimized as an Lck kinase inhibitor), as Novartis. They started their project by screening against Lck, several analogs of a hit found by HTS for another project (TNF-α), and having a trisubstituted urea structure.

articles
Development of N-2,4-pyrimidine-N-phenyl-N'-phenyl ureas as inhibitors of tumor necrosis factor alpha (TNF-a) synthesis. Part 1 Todd A. Brugel, Jennifer A. Maier, Michael P. Clark, Mark Sabat, Adam Golebiowski, Roger G. Bookland, Matthew J. Laufersweiler, Steven K. Laughlin, John C. VanRens, Biswanath De, Lily C. Hsieh, Marlene J. Mekel and Michael J. Janusz Bioorganic and Medicinal Chemistry Letters 16 2006
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Optimization Towards Lck Kinase Inhibition¶
A hit with a modest inhibitory activity (IC50 = 3.86 µM) was found. The systematic modification of the structure of this initial hit led to the preparation of about 40 analogs. Low nanomolar inhibitors of the Lck protein kinase were found, one of them corresponded exactly to the Novartis molecule 1.

articles
Development of N-4,6-pyrimidine-N-alkyl-N0-phenyl ureas as orally active inhibitors of lymphocyte specific tyrosine kinase Jennifer A. Maier, Todd A. Brugel, Mark Sabat, Adam Golebiowski, Matthew J. Laufersweiler, John C. VanRens, Corey R. Hopkins, Biswanath De, Lily C. Hsieh, Kimberly K. Brown, Vijayasurian Easwaran and Michael J. Janusz Bioorganic and Medicinal Chemistry Letters 16 2006
Summary¶
Based on modeling considerations, Novartis designed a mimic of a pyrido[2,3-d]pyrimidin-7-one scaffold (PD-166285) by moving the nitrogen atom and replacing the pyrimidone ring by a urea moiety, in order to form an intramolecular hydrogen bond that preserved the molecular geometry unchanged. The molecule was synthesized and proved to inhibit several protein kinases at the nanomolar level. At the same time, the same molecule was discovered independently by Procter & Gamble, based on the systematic modification of a structure discovered by HTS.

articles
Entry into a new class of protein kinase inhibitors by pseudo ring design P. Furet, G. Caravatti, V. Guagnano, M. Lang, T. Meyer and J. Schoepfer Bioorganic and Medicinal Chemistry Letters 18 2008
Case Study-3 : Anthranilamide Scaffold¶
Anthranilamide Scaffold¶
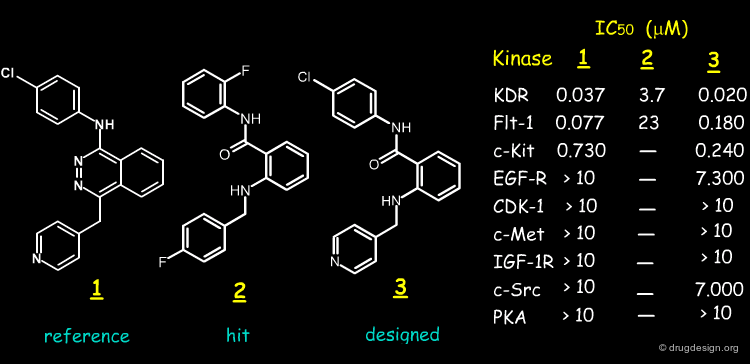
Here we present the story of the design of a new type of structure by P. Furet at Novartis and created through modeling and scaffold morphing. The story starts with the discovery of the anilinophtalazine compound 1. The molecule was discovered by HTS and found to be a potent KDR and Flt-1 protein kinase inhibitor. It was subjected to clinical trials under the name of PTK-787.

articles
New anilinophthalazines as potent and orally well absorbed inhibitors of the VEGF receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis Bold, G.; Altmann, K.-H.; Frei, J.; Lang, M.; Manley, P. W.; Traxler, P.; Wietfeld, B.; Brueggen, J.; Buchdunger, E.; Cozens, R.; Ferrari, S.; Furet, P.; Hofmann, F.; Martiny- Baron, G.; Mestan, J.; Roesel, J.; Sills, M.; Stover, D.; Acemoglu, F.; Boss, E.; Emmenegger, R.; Laesser, L.; Masso, E.; Roth, R.; Schlachter, C.; Vetterli, W.; Wyss, D.; Wood, J. M. J. Med. Chem 43 2000
** ** Bold, G.; Frei, J.; Furet, P.; Manley, P. W.; Bruggen, J; Cozens, R.; Ferrari, S.; Hofmann, F.; Martiny-Baron, G.; Mestan, J.; Meyer, T.; Wood, J. M. Drugs of the Future 27 2002
Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA J. Med. Chem 39 1996
Structural Determinants of Anilinophtalazine Activity ?¶
When the anilinophtalazine lead was discovered, nothing was known about the structural determinants behind its good biological activities. The Novartis team addressed the following questions: what is the bioactive conformation of the molecule and what is the importance of the two nitrogen atoms of the phtalazine heterocycle?

articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA J. Med. Chem 39 1996
Conformational Analyses¶
To determine the nature of the bioactive conformation, a conformational analysis study was carried out by Novartis modelers to assess the preferred conformation of the molecule. The calculations indicated two possible orientations of the aniline moiety (syn and anti), with respect to the phtalazine nucleus in 1. The "anti" conformation was preferred and the difference in energy between the "syn" and "anti" conformers was 3.1 kcal/mol (populations 77% and 23%).

articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Bidentate Binding Mode Unlikely to Occur¶
Experimental and modeling studies reveal that most protein kinase inhibitors bind to the hinge region of the enzyme involving H-bond donor/H-bond acceptor bidentate binding. The Novartis team saw that the anilinophtalazine could bind according to this classical mode if the molecule took on the syn conformation; however this form has relatively high energy. They concluded that it would be more advantageous to pursue analyses with the "anti" conformation.
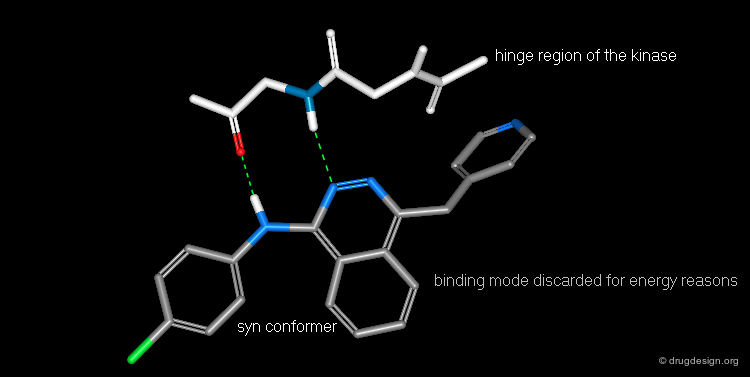
articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Role of the Nitrogen Phtalazine Atoms¶
If the bioactive conformation of 1 is likely to be "anti", this means that the anilino NH and the H-bond donor/H-bond acceptor system are not involved in the bidentate binding to the kinase. This prompted the Novartis team to explore the role of the two nitrogen atoms of the phtalazine heterocycle in greater depth.

Database Searching¶
A database search was undertaken to identify similar structures lacking one or both phtalazine nitrogen atoms, but no hits were found. Another hypothesis was tested by the Novartis team by mimicking the pyridazine ring of phtalazine by the pseudo-cycle of a structure possessing an intramolecular hydrogen bond. A search in the Novartis corporate database using the anthranilic acid amide query structure indicated below (second button) gave one single hit. This compound was tested biologically and showed good activity for KDR and Flt-1 kinase inhibition.


articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
3D Electrostatic Potential¶
A study was made to compare the electrostatic potential on the surfaces of phtalazine and anthranilic acid amide model molecules (button "2D"). It showed a high degree of mimicry between the two systems.
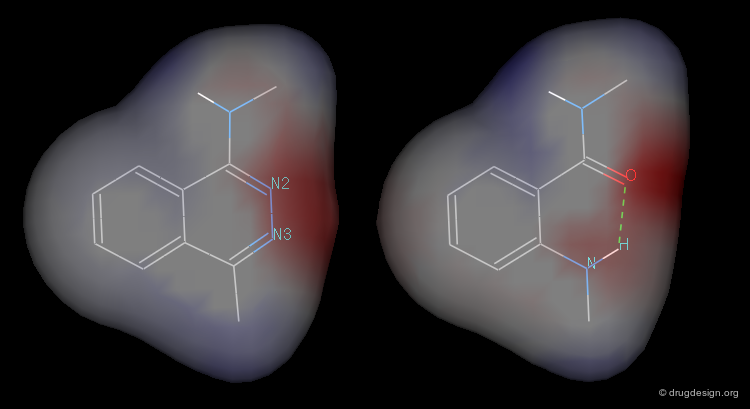

articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA J. Med. Chem 39 1996
Synthesis of the Exact Anthranilamide Mimetic¶
The good biological results obtained for the hit prompted the synthesis of the exact anthranilic amide mimetic of 1 (molecule 3). The synthetic scheme used is described below. Note that this is a nice illustration of database searching conducted solely to validate a concept.


articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA J. Med. Chem 39 1996
Biological Tests¶
The biological tests made on molecule 3 revealed that the compound is a potent inhibitor of KDR/Flt-1, with similar potency and selectivity as compared to 1.
articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
3D Overlay of Mimic Structures¶
The good biological activities found for molecule 3 confirmed the validity of the hypotheses considered for its design. The most important is the intramolecular H-bond between the aniline N-H and the benzanilide C=O that favors the conformation which mimics the reference bicyclic anilinophtalazine structure. The oxygen atom of the carbonyl group of the benzanilide mimics the N2 nitrogen atom of the phtalazine.


articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Tyrosine kinase inhibitors. 8. An unusually steep structure-activity relationship for analogues of 4-(3-bromoanilino)-6,7-dimethoxyquinazoline (PD 153035), a potent inhibitor of the epidermal growth factor receptor Bridges AJ, Zhou H, Cody DR, Rewcastle GW, McMichael A, Showalter HD, Fry DW, Kraker AJ, Denny WA J. Med. Chem 39 1996
Determinants for Anilinophtalazine KDR/Flt-1 Activities¶
These results suggested the following determinants for anilinophtalazine KDR/Flt-1 kinase inhibitory activities: (1) the bioactive conformation of the anilinophtalazine structure is the "anti" conformation; (2) the nitrogen atom N2 is important whereas (3) the nitrogen atom N3 is not important.

articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Summary¶
Theoretical analyses made on an anilinophtalazine HTS lead 1 and exploited by an intelligent design were followed up by a database search that enabled the identification of an anthranilamide chemotype kinase inhibitor hit. This prompted the synthesis of the exact mimic of the initially designed molecule 3; it proved to be active at the nanomolar level. The design was made with structure-based reasoning and the work required the biological test of only one hit and the synthesis of only one compound !

articles
Identification of a new chemical class of potent angiogenesis inhibitors based on conformational considerations and database searching Furet P, Bold G, Hofmann F, Manley P, Meyer T, Altmann KH Bioorg. Med. Chem. Lett 13 2003
Case Study-4 : Phenoxyphenyltriazoles¶
Phenoxyphenyltriazoles¶
Akbarzadeh et al. worked to discover novel benzodiazepine (BZD) ligands with simple non-rigid structures that differed from those of diazepam and estazolam.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Requirements for Binding to the BZD Receptor¶
Diazepam 1 and estazolam 2 are potent benzodiazepine receptor antagonists. Structure-activity relationships of benzodiazepine ligands indicate that the following two features are needed for binding to the benzodiazepine receptor: (1) an aromatic ring, and (2) a coplanar proton-accepting group at the proper distance. Moreover, it has been shown that a second out-of-plane aromatic ring can provide additional potency for binding to the receptor.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Design of an Estazolam Mimic¶
Based on the requirements mentioned in the previous page, and using estazolam 2 as a reference lead, Akbarzadeh et al. designed structures such as 3 as possible benzodiazepine ligands.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Conformational Analyses and Overlay with Diazepam¶
The conformational analysis of 3 was made, and the preferred conformer was superimposed on that of estazolam. The overlay shows a good match between the two structures. The designed structure has an intramolecular hydrogen bond that contributes to maintaining the molecule in a geometry comparable to that of estazolam.


articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Chemical Synthesis of the Mimics¶
About 16 Phenoxyphenyltriazole derivatives were synthesized. Analog 3 was prepared according to the scheme shown below.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Confirmation of the Design Hypothesis¶
The 16 Phenoxyphenyltriazole derivatives were pharmacologically tested and it was found that compound 3 has activities similar to that of diazepam on the rotarod test. This confirms the validity of the design hypothesis. The structure has an intramolecular hydrogen bond that contributes to maintaining the molecule in a geometry comparable to that of estazolam.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Summary¶
Based on proposed SAR and using estazolam as a reference, a simple non-rigid structure such as 3 was designed and proved to have a benzodiazepine activity comparable to diazepam on the rotarod test. The designed structure has an intramolecular hydrogen bond that contributes to maintaining the molecule in a geometry comparable to that of estazolam.

articles
Design and Synthesis of 4H-3-(2-Phenoxy)phenyl-1,2,4-triazole Derivatives as Benzodiazepine Receptor Agonists Akbarzadeh T, Tabatabai SA, Khoshnoud MJ, Shafaghic B and Shafiee A Bioorganic and Medicinal Chemistry 11 2003
Case Study-5 : Pro-Leu-Gly-NH2 Peptide¶
Pro-Leu-Gly-NH2 Peptide¶
The Pro-Leu-Gly-NH2 (molecule 1) is a dopamine receptor-modulating peptide held by an intramolecular hydrogen-bond that stabilizes the molecule in a "C5" conformation, characterized by a pseudo-5-membered ring. The structure of this peptide was transformed by scaffold morphing, as explained in the following pages.
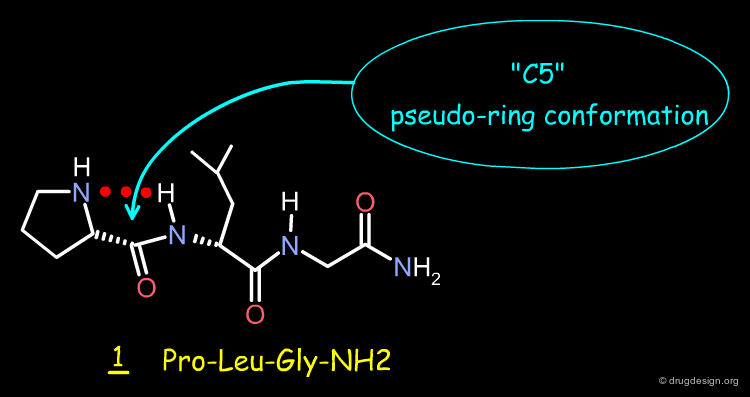
The γ-Lactam Analog of Pro-Leu-Gly-NH2¶
The existence of a "C5" conformation in the Pro-Leu-Gly-NH2 peptide is evidenced by the X-ray structure of the γ-lactam analog 2, which adopts a "C5" conformation, This molecule was found to be 1000 times more potent than the initial Pro-Leu-Gly-NH2 peptide in enhancing the binding of N-propylapomorphine to dopamine D2 receptors.


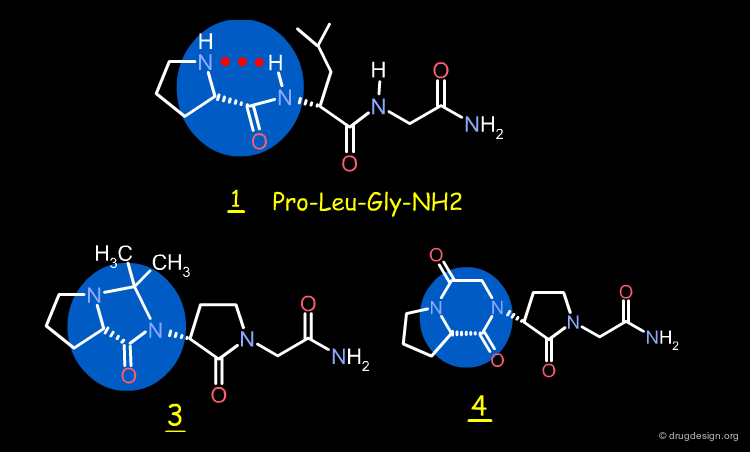
Design of Imidazolidinone and Diketopiperazine¶
Two rigid analogs of 2, the imidazolidinone 3 and the diketopiperazine 4, were synthesized as potential mimics of the "C5" conformation of 2 by replacing the pseudo-ring by a real one.

Biological Tests¶
The biological studies showed that 3 and 4 modulate high-affinity dopamine D2 receptors. The pharmacological activities of the two molecules were found to be of the same magnitude, and 1000 times more potent than the Pro-Leu-Gly-NH2 peptide.

3D Alignment of Pro-Leu-Gly-NH2 and Mimics¶
The 3D superimposition of the Pro-Leu-Gly-NH2 peptide 1 with the imidazolidinone and diketopiperazine analogs 3 and 4 is shown below and demonstrates the good biological activities observed for these synthetic analogs.

Summary¶
The two synthetic molecules 3 and 4 were derived by Baures et al. to mimic the bioactive conformation of the dopamine receptor-modulating peptide Pro-Leu-Gly-NH2 (molecule 1). This bioactive conformation is characterized by a "C5" conformation that is stabilized by an intramolecular hydrogen bond. The rigid molecules proved to be very active in the modulation of dopamine D2 receptors.
Case Study-6 : Remoxipride Mimic¶
Remoxipride Mimic¶
Desmethylremoxipride (FLA-797, molecule 2) is the demethylated analog of remoxipride 1, an antagonist of the dopamine type 2 receptor developed for treating schizophrenia. The structure of this molecule was transformed by scaffold morphing, as explained in the following pages.

articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents M.H. Norman, J.L. Kelly and E.B. Hollingsworth J Med Chem 36 1993
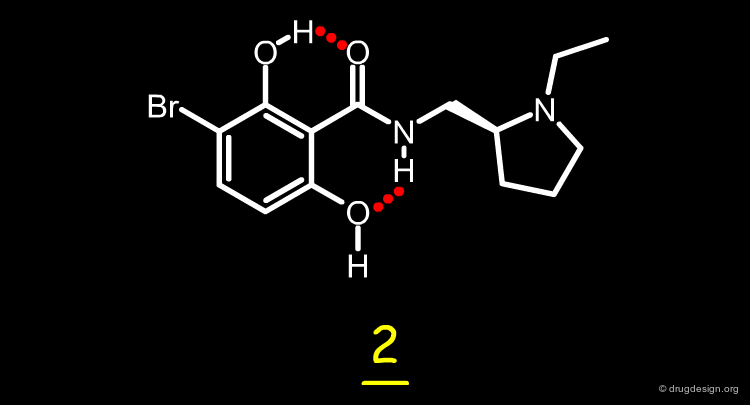
Bioactive Conformation of Desmethylremoxipride¶
The high in-vitro biological activity of desmethylremoxipride (IC50 = 12 nM) was attributed to the presence of a planar arrangement between the amide carbonyl and the aromatic ring. The planarity is maintained by an intramolecular hydrogen bonding framework, as illustrated in the figure.
articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents M.H. Norman, J.L. Kelly and E.B. Hollingsworth J Med Chem 36 1993
Design of Rigid Analog¶
A conformationally restricted phtalimidine molecule 3 was designed which contains a covalent connection between the amide nitrogen and the ortho position of the aromatic ring.

articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents M.H. Norman, J.L. Kelly and E.B. Hollingsworth J Med Chem 36 1993
Chemical Synthesis¶
The synthesis of the phtalimidine molecule 3 is described below.

articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents M.H. Norman, J.L. Kelly and E.B. Hollingsworth J Med Chem 36 1993
Biological Tests¶
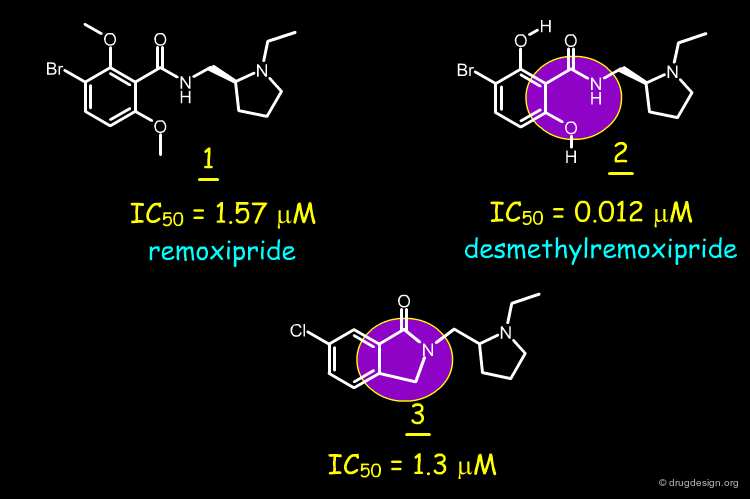
Remoxipride 1 has a moderate affinity for the dopamine D-2 receptor in vitro (IC50 = 1.57 µM). The desmethyl analog 2 with its two intramolecular hydrogen bonds maintaining the system coplanar, is more potent (IC50 = 0.012 µM). The phtalimidine compound 3 has a potency comparable to that of remoxipride 1 (IC50 = 1.3 µM). About 15 analogs were synthesized, however compound 3 remained the most potent.

articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents M.H. Norman, J.L. Kelly and E.B. Hollingsworth J Med Chem 36 1993
3D Alignments¶
The superimposition of desmethylremoxipride 2 and isoindolinone 3 is shown in the view below.
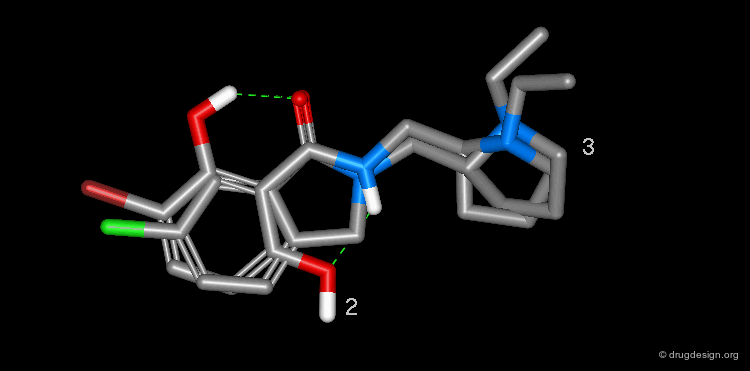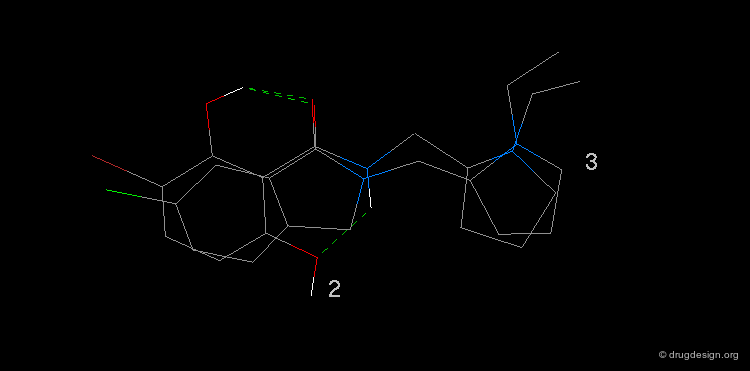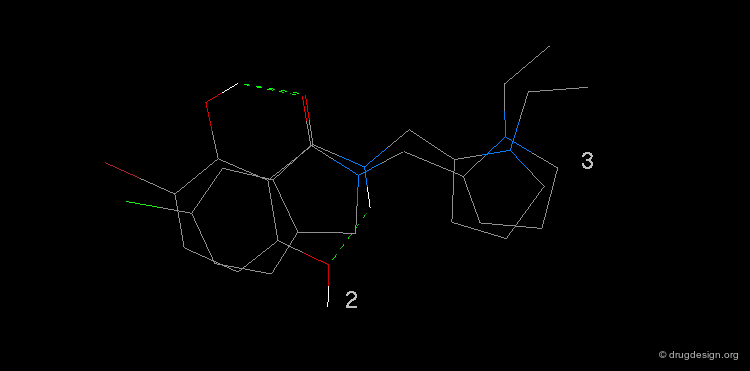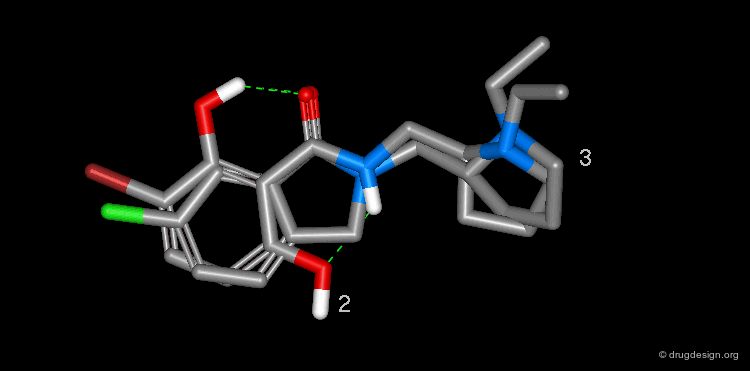
Summary¶
In order to find new antipsychotic agents, the phtalimidine molecule 3 was designed to mimic one intramolecular hydrogen bond found in 2. The compound was found to be as potent as remoxipride 1 as a dopamine D-2 antagonist, but less potent than its desmethylremoxipride analog 2.
articles
Conformationally restricted analogues of remoxipride as potential antipsychotic agents M.H. Norman, J.L. Kelly and E.B. Hollingsworth J Med Chem 36 1993
Case Study-7 : Rimonabant Mimics¶
Rimonabant Mimic¶
Rimonabant 1 is an antagonist of the cannabinoid type 1 receptor (CB-1) used for the treatment of obesity. The structure of Rimonabant was transformed by scaffold morphing, as explained in the following pages.

articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
Conformational Analysis of Rimonabant¶
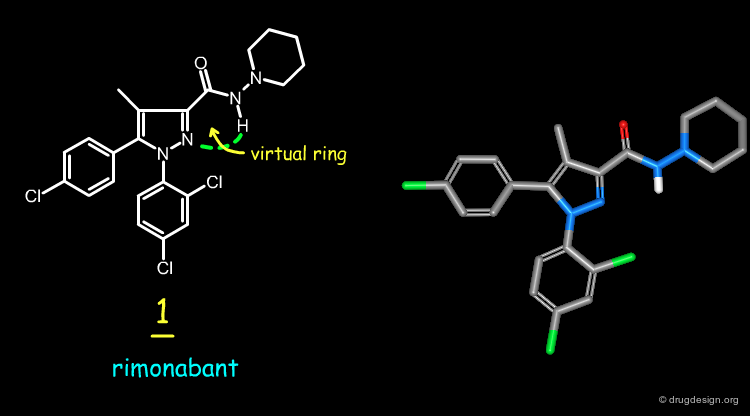
Computational studies indicate that the preferred conformation of rimonabant has a trans-amide with the carboxamide oxygen atom nearly coplanar with the pyrazole ring and oriented in the same direction as the pyrazole C-4 methyl group. Subsequent X-ray studies were made on 1 and confirmed the computational predictions.
articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) interaction with LYS 3.28(192) is crucial for its inverse agonism at the cannabinoid CB1 receptor Hurst DP, Lynch DL, Barnett-Norris J, Hyatt SM, Seltzman HH, Zhong M, Song ZH, Nie J, Lewis D, Reggio PH. Mol Pharmacol 62(6) 2002
Design of Rigid Analog¶
Analog 2 was designed by replacing the pyrazole N-2 of rimonabant by a carbon atom, and making a six-membered ring between this atom and the carboxamide nitrogen, to freeze the geometry of the molecule. The docking of 1 to a model of the CB-1 receptor shows that the carboxamide nitrogen atom is not involved in a hydrogen-bond with the receptor, and this supports the idea of designing a constrained analog.

articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
Chemical Synthesis¶
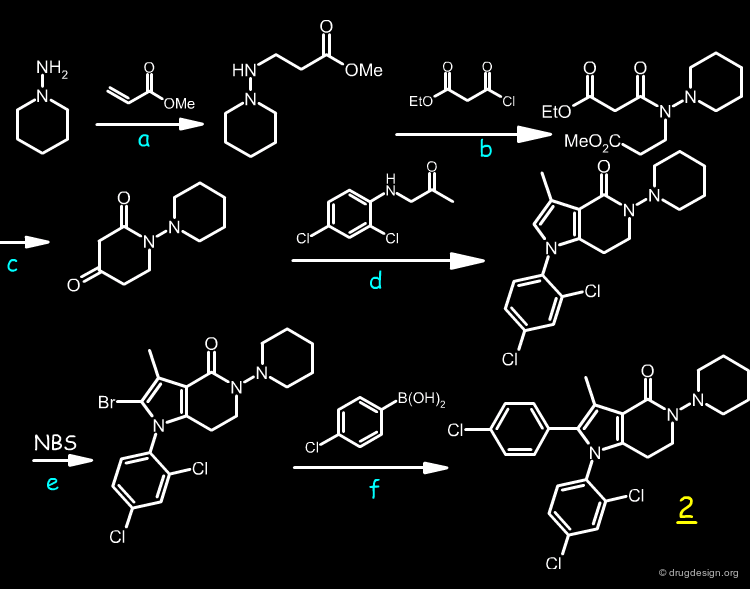
The synthesis of 2 involved six steps, as described below.
articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
Biological Tests¶
Compound 2 proved to be a very potent antagonist of the cannabinoid type 1 receptor, like rimonabant (molecule 1). Additional studies indicated that it caused appetite suppression and reduction in body weight gain in rat models used to develop new therapeutics for obesity.

articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
3D Alignment of Rimonabant and Mimic¶
The view below shows the good superimposition of the 3D structure of compound 2 with that of rimonabant 1.

articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
Summary¶
The rigid analog 2 of rimonabant 1 is a potent antagonist of the cannabinoid type 1 receptor. The virtual ring found in the rimonabant structure is mimicked by a rigid 6-membered piperidinone ring.

articles
Constrained analogs of CB-1 antagonists: 1,5,6,7-Tetrahydro-4H-pyrrolo[3,2-c]pyridine-4-one derivatives Smith RA, Fathi Z, Brown SE, Choi S, Fan J, Jenkins S, Kluender HC, Konkar A, Lavoie R, Mays R, Natoli J, O Connor SJ, Ortiz AA, Podlogar B, Taing C, Tomlinson S, Tritto T, Zhang Z. Bioorg Med Chem Lett 17(3) 2007
Case Study-8 : Salicylamide Mimics¶
Salicylamide Mimics¶
Salicylamide 1 is a competitive inhibitor of the scytalone dehydrase. Hodge and Pierce exploited its structure to design potent mimics of this inhibitor. The structure of this molecule was transformed by scaffold morphing, as explained in the following pages.

articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
SAR of Salicylamide 1¶
Analysis of the SAR data of the salicylamide series provides useful information about the structural requirements for the binding of 1.

articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
Removing the Hydroxyl or the Carbonyl¶
It has been observed that the deletion of either the phenolic OH or the carbonyl oxygen atom dramatically reduces scytalone dehydrase inhibitory activity. These two functional groups are necessary to good inhibitory activity.

articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
Analyzing if Ortho Electron Lone-Pair is Sufficient¶
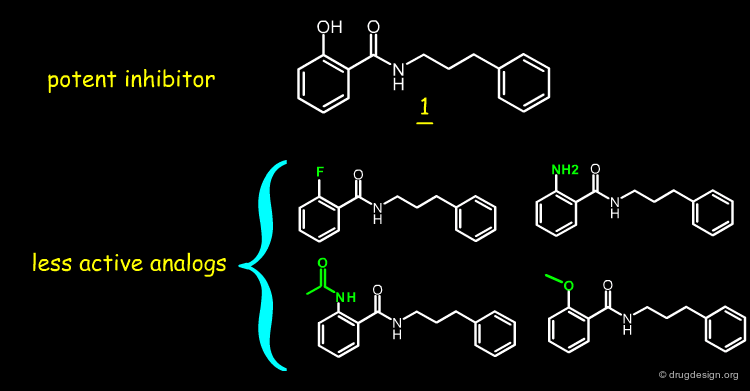
When the hydroxyl of 1 is replaced by a fluoro, amino, acetamido or methoxy, the inhibitory activity decreases by more than two orders of magnitude. This indicates that an electron lone-pair at the ortho-position is insufficient to explain the observed activities and the hydroxyl group is necessary for good inhibitory activity.
articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
Potent Inhibition at Ki at Different pH¶
The affinity of 1 against the scytalone dehydrase was measured at different pH. The authors found that the neutral form of 1 binds 30-fold more tightly to the enzyme than the deprotonated anionic form. This once again demonstrates the importance of the phenolic hydroxyl group; moreover the OH needs to be in its neutral form.

articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
Pseudo-Ring of 1 Binds as a Whole Unit¶
Hodge and Pierce analyzed all the data presented in the previous pages and concluded that these SAR are consistent with the idea that the pseudo-ring of 1 binds as a "whole intact block".

articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
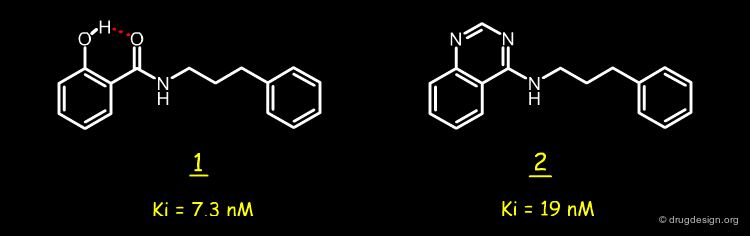
Design of Quinazoline Mimic¶
With the aim of stabilizing the salicylamides to metabolic deactivation, Hodge and Pierce imagined the replacement of the pseudo-ring of 1 by the pyrimidine ring of a quinazoline molecule such as 2. Through kinetic studies, they demonstrated that the two inhibitors 1 and 2 have similar potencies and bind to the enzyme in their neutral form. Removing one of the two nitrogen atoms in the quinazoline structure 2 yields molecules such as 3 and 4 that are much less active.
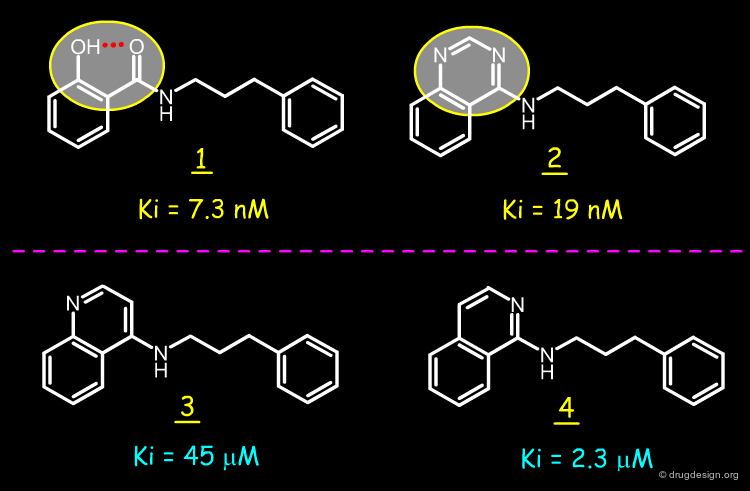
articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
3D Alignment of Salicylamide 1 and Quinazoiline 2¶
The 3D alignment of the salicylamide 1 and quinazoline 2 indicates an excellent match for the two structures.

Conclusion¶
The results shown here suggest that the scytalone enzyme recognizes the pseudo-ring of salicylamide 1 as a whole intact block. This observation might be general and valid for other proteins.

articles
A diazine heterocycle replaces a six-membered hydrogen-bonded array in the active site of Scytalone Dehydratase C.N. Hodge and J. Pierce Bioorganic and Medicinal Chemistry Letters 3 (8) 1993
Summary¶
Based on the structure of salicylamide 1 and its associated SAR data, Hodge and Pierce derived a quinazoline mimic 2 that proved to be a potent inhibitor of the scytalone dehydrase, active at the nanomolar range.
Case Study-9 : Bradykinin Antagonists¶
Bradykinin Antagonists¶
Involved in the design of bradykinin antagonists, the Merck group needed to find a replacement for a 2,3-diaminopyridine moiety in one of their most potent compounds. Based on conformational analyses, molecular mimicry and bioisosterism, the 2,3-diaminopyridine moiety was progressively replaced by an improved moiety, as explained in the following pages.
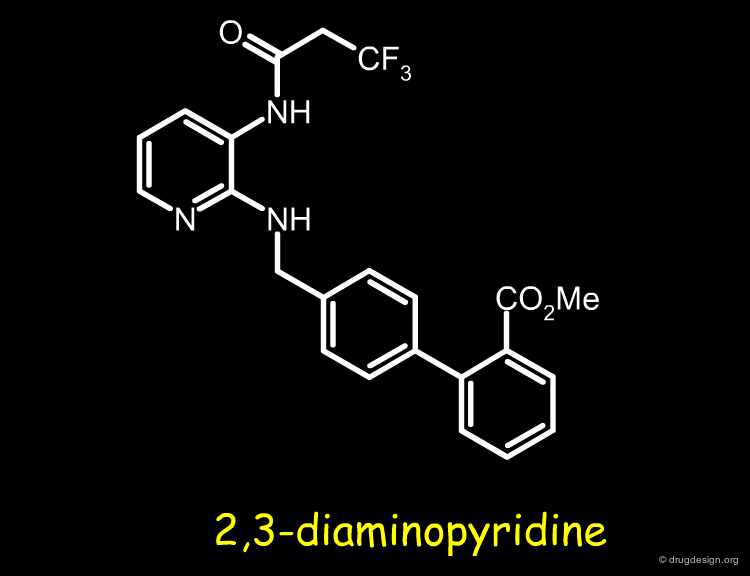
articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
The Problem¶
Molecule 1 was found to be a potent bradykinin B1 antagonist but could not be considered for further development because of the toxicity of the metabolites associated with the diaminopyridine fragment. It was important to find a replacement for this moiety in order to overcome this problem.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
The Stepwise Discovery of Cyclopropylamide¶
The design strategy followed a stepwise process that is described in detail here.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Retaining the two N-H groups¶
Analogs of 1 and the resulting SAR data indicated that it was necessary to retain the two N-H bonds, the N-acyl group, and its geometrical proximity to the biphenyl region. A simple way to do so was to connect the N-acyl group and the biphenylamine with an ethylene linker (prototype A).

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Mimicking the Nitrogen Pyridine Atom by a Carbonyl¶
It was also imperative to retain a heteroatom in the region occupied by the nitrogen atom of the pyridine. A carbonyl group was therefore introduced to mimic the nitrogen atom and ensure potential lone pair interactions (prototype B). Moreover, the carbonyl was introduced to reduce the basicity of the benzylic amine in A to achieve closer resemblance to the diaminopyridine structure.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Conformational Considerations¶
Because analog B was unlikely to adopt a conformation with a molecular geometry similar to that of 1, the Thorpe-Ingold effect was considered, leading to the possibility of adding a gem-dimethyl group at the C alpha position (prototype C) to facilitate the rotation about the CO-C alpha bond and increase the population of the target conformer mimic.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
First Molecules Synthesized¶
The reasoning explained in the previous page led to the synthesis of a molecule such as 2 as a first replacement for the 2,3-diaminopyridine moiety. This molecule had a modest inhibitory activity (IC50=3.45 µM); whereas analog 3 was as expected much less active.
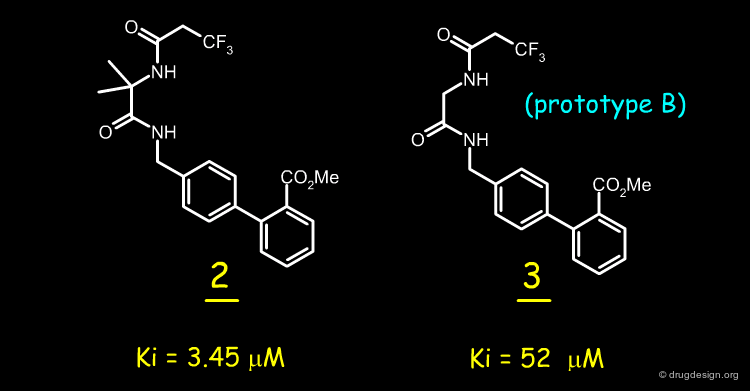
articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Restoring Lipophilic Interactions¶
The comparison of the structures of 1 and 2 suggested that lipophilic functionalities at the C alpha atom could be exploited to mimic the lipophilicity of the pyridine fragment. The cyclohexylamino acid analog 6 was prepared and showed improved binding affinity as compared to 2.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Reducing Ring Size¶
Analogs having a ring size of 5, 4 and 3-membered atoms were also prepared and tested. It was found that the smaller the size of the ring, the better the binding affinity.
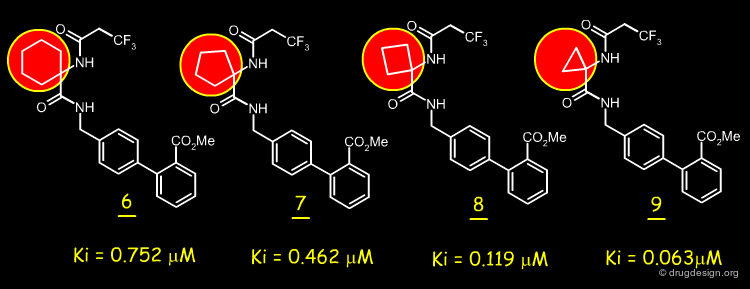
articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
The Best Replacement¶
The cyclopropylcarbonyl analog 9 proved to correspond to the best surrogate of the 2,3-diaminopyridine moiety. Variations at the N-acyl group enabled the researchers to find more potent inhibitors such as 10.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Additional Factors in Cyclopropyl Replacement¶
The cyclopropyl analog has additional advantages that are discussed here.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Torsion Angle N-C-C-N¶
In order to mimic the molecular geometry of 1, structure C must have a torsion angle Φ(N-C-C-N) close to 0°. This is evidenced by the low activity of 3 (preferred torsion angle of 180°) and the higher activity of 2 (preferred gauche torsion angle).

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Smaller Rings have Increasing π Character¶
Another factor that explains the observation of increased activities when reducing the size of the ring is that smaller rings have a higher p character (cyclopropyl groups are often seen as equivalent to a double bond). Therefore, there are π-π overlaps between the carbonyl and the cyclopropyl groups, an overlap that is maximal when the two groups are coplanar. This constitutes an additional reason why the cyclopropylamino acid amide group is a good pharmacophoric replacement for the 2,3-diaminopyridine.
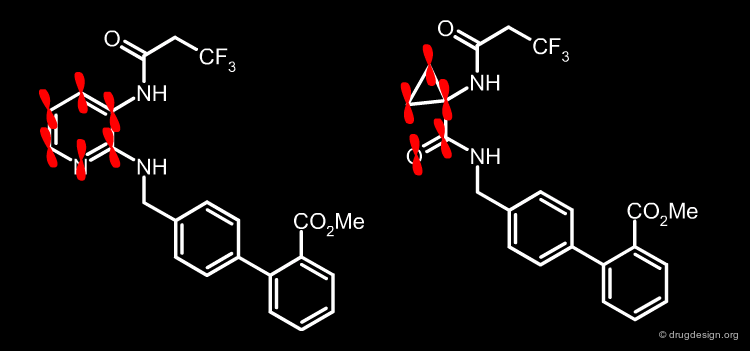
articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Ring Strain and Geometry of Cyclopropyl¶
The ring strain existing in the cyclopropyl ring contributes to increasing the mimicry with the diaminopyridine fragment. The explanation is the following: the bond angle between the geminal substituents becomes closer to 116° (Thorpe-Ingold effect), instead of the normal tetrahedral value 109°. This increases the geometrical similarity with the sp2 bond angles of 120° found in the diaminopyridine core.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Bulkiness of the Hydrophobic Ring¶
Another reason enabling cyclopropyl analog 9 to adopt a conformation more similar to 1 more easily is the bulkiness of the ring. With a 3-membered ring, the steric effects are minimal and enable the N-acyl group to adopt a conformation similar to that found in 1.
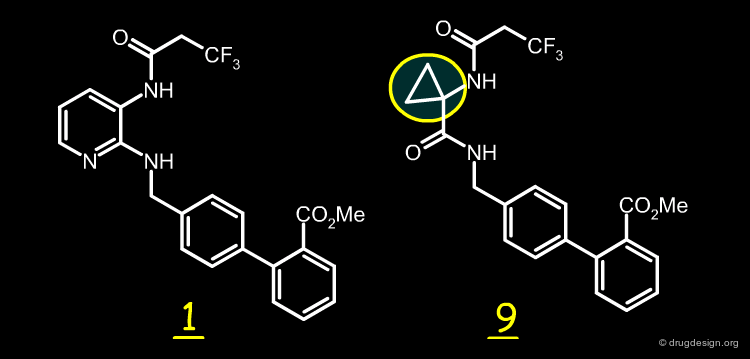
articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
3D Alignment of 1 and the Cyclopropyl Surrogate¶
Below is shown the good 3D overlap of the reference diaminopyridine structure with that of the cyclopropylamino acid amide. This explains why the cyclopropylamino acid amide is a good surrogate of the 2,3-diaminopyridine moiety.

Summary¶
To circumvent toxicity problems of a 2,3-diaminopyridine bradykinin antagonist, the Merck group designed a cyclopropylamino acid amide surrogate. To achieve this good mimicry, the group took into account the geometric, conformational, stereoelectronic and physicochemical features of the structures. The antagonists displayed low nanomolar affinity for the human bradykinin receptor and good oral bioavailability in the rat.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Factor Xa Inhibitors¶
Confronted with the same problem encountered for bradykinin antagonists, the Merck group had very potent Factor Xa inhibitors carrying a 2,3-diaminopyridine moiety that could not be further developed.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Factor Xa Inhibitors with 2,3-Diaminopyridine Core¶
After the good results obtained with bradykinin antagonists, the Merck team envisaged replacing the 2,3-diaminopyridine moiety by a cyclopropylamino acid amide, to see whether this replacement could also be valid for this project. The replacement of the 2,3-diaminopyridine in molecule 12 by a cyclopropyl acid amide yielded compound 13 with a Ki of 175 nM.
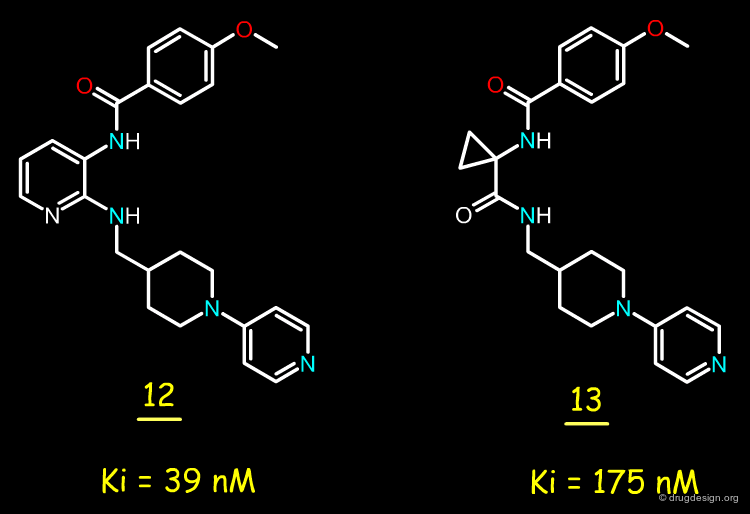
articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Replacement May be of General Utility¶
This result suggests that the replacement of 2,3-diaminopyridine by a cyclopropylcarbonyl moiety may be of general utility and valid for other targets.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
Surrogates Generated by Computer¶
It is interesting to note that Paul Hawkins (OpenEye Scientific Software Inc.) used the Brood program to find bioisoteres of 2,3-diaminopyridine and found the same replacement as discovered by the Merck team. Some of the hits generated by the program are represented below. Note that cyclopropylcarbonyl is ranked in position 4. Other programs also exist that can be used along the same lines (see section on bioisosterism) and be exploited for scaffold morphing.

articles
Cyclopropylamino acid amide as a pharmacophoric replacement for 2,3-diaminopyridine. Application to the design of novel bradykinin B1 receptor antagonists Wood MR, Schirripa KM, Kim JJ, Wan BL, Murphy KL, Ransom RW, Chang RS, Tang C, Prueksaritanont T, Detwiler TJ, Hettrick LA, Landis ER, Leonard YM, Krueger JA, Lewis SD, Pettibone DJ, Freidinger RM, Bock MG. J Med Chem 49(4) 2006
OpenEye Scientific Software Inc P. Hawkins
Personnal communication
Similarities in bioanalogous structural transformation patterns among various bioactive compound series Fujita T Biosci. Biotech. Biochem 60(4) 1996
Bioisosteric morphing in primary hit optimization KV Balakin, SE Tkachenko, I Okun, AV Skorenko, YA Ivanenkov, NP Savchuk, AA Ivashchenko and Y Nikolsky Chimica Oggi - Chemistry Today Jan-Feb 2004
Extended Summary: BIOSTER - a database of structurally analogous compounds Ujvary I Pesticide Science 51 (1) 1997
The quest for bioisosteric replacements M Wagener and JPM Lommerse J Chem Inf Model 46(2) 2006
Drug rings database with web interface. A tool for identifying alternative chemical rings in lead discovery programs Lewell XQ, Jones AC, Bruce CL, Harper G, Jones MM, McLay IM, Bradshaw J. J Med Chem 46(15) 2003
COSMOsim: bioisosteric similarity based on COSMO-RS sigma profiles Thormann M, Klamt A, Hornig M, Almstetter M. J Chem Inf Model 46(3) 2006
book
Toshio Fujita Trends in QSAR and Molecular Modeling 92. Procedings of the 9th European Symposium on Structure-Activity Relationships: QSAR and Molecular Modelling ESCOM - Leiden 1993
Toshio Fujita et al. Kyoto University and EMIL PROJECT, Fujitsu Kansai Systems Laboratory, Osaka, Japan QSAR and Drug Design: New developments and applications Elsevier, Pharmacochemistry Library Volume 23, H. Timmerman Ed. 1995
Toshio Fujita ACS Symposium series No. 606 Classical and Three-Dimensional QSAR in Agrochemistry The American Chemical Society 1995
ADDITIONAL CASE STUDIES¶
Additional Case Studies¶

Copyright © 2024 drugdesign.org