Case Studies in 3D Database Searching¶
Info
Successful examples of 3D database searching are presented and discussed. They show how 3D searching can be used either alone, or in combination with structure-based searching to discover hits with a good potential. These examples demonstrate that when good models are developed 3D database searching can lead to the rapid discovery of lead compounds with a minimum of additional synthetic chemical effort.
Number of Pages: 80 (±2 hours read)
Last Modified: July 2008
Prerequisites: 3D Database Searching
Case Study-1 : Ligands of the Dopamine D3 Receptor¶
Reference Compounds¶
To discover novel ligands of the dopamine-3 (D3) subtype, a hybrid approach was developed based on stepwise pharmacophore and structure-based searching. The following ten known D3 ligands were selected as a starting point for the development of the pharmacophore model.

Pharmacophore Model¶
Chemical structural analyses indicated that the D3 ligands contained a common aromatic ring and a nitrogen atom sp3 connected to a propyl group, which could be superimposed in 3D.


Model of the Dopamine D3 Receptor¶
A model of the 3D structure of the D3 receptor was constructed by homology with the high-resolution crystallographic structure of rhodopsin. This model was further refined by molecular dynamic treatments, in the presence of the lipid-water environment.

Residues Involved in the Binding¶
Further docking studies identified the residues involved in the binding of the ligands. The following view illustrates the predicted binding mode of R-(+)-7-OH-DPAT (ref-2), one of the most well characterized D3 partial agonists to date.

Combined Pharmacophore and Structure-Based Searching¶
Initially the pharmacophore search generated a great number of hits. These hits were then subjected to structure-based screening to identify compounds that had effective interactions with the receptor. Finally, top-ranked compounds were defined as those with structural novelty, based on comparisons with the known D3 ligands. The most promising structures were selected for experimental biological testing.

Results of the 3D Searching¶
Using the NCI 3D database of 250,200 "open" compounds, 6727 hits were identified by the 3D pharmacophore screening, 2478 successfully passed the structure-based screening and 1314 of them remained after the structural novelty screening. Twenty of them were selected for testing their activities: four of them were found with Ki values between 11 and 63 nM. Seven others were found with sub-µM activities.
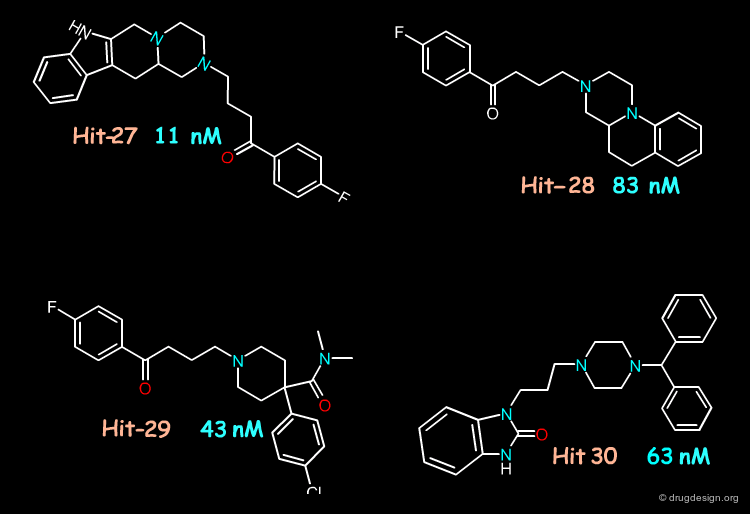

Summary¶
The approach presented here combined pharmacophore and structure-based searching, which helped discover potent ligands of the D3 receptor. The molecules shown below were discovered as hits that existed in the database; they did not require the development of a synthetic program. This type of methodology may be more effective than using 3D pharmacophore screening alone.

Browser of Dopamine D3 Receptor Ligands¶

Case Study-2 : Non-Peptidic Cyclophilin Ligands¶
Reference Compound: Cyclosporin A¶
Cyclosporin-A is a clinically important immunosuppressant drug that binds to immunophilin binding proteins such as cyclophilin-A. A 3D pharmacophore-based approach has been devised for purposes of discovering non-peptidic cyclophilin ligands.

The Bioactive Conformation of Cyclosporin A¶
The X-ray structure of cyclosporin-A (in light blue) bound to cyclophilin-A (a few residues are displayed in brown) was available, which provided information on the bioactive conformation of the ligand.

Pharmacophore Model¶
Based on analyses of the X-ray data that indicated which interactions are exploited by cyclosporin-A when it binds to cyclophilin-A, a pharmacophore model was derived and consisted of a hydrophobic moiety, and five hydrogen bond interactions involving five successive residues of cyclosporin-A.


Results of 3D Searching¶
Using the query described in the previous page, a pharmacophore search was initiated with three 3D-databases (Available Chemical Database, World Drug Index, and Chapman-Hall Dictionary of Organic Compounds). Molecules that had the desired hydrophobic fragment, while simultaneously forming at least three of the five hydrogen bonds previously defined, were selected as hits. The following structure proved to inhibit cyclophilin rotamase with an IC50 of 5.9 µM, and was selected as a lead for subsequent SAR.

Superposition of the Hit with Cyclosporin-A¶
The following view illustrates the superposition of the lead compound (in yellow) with the structure of cyclosporin-A. Some residues of the cyclophilin protein are also included (in brown); they visualize the hydrogen bond interactions considered in the 3D pharmacophore model.
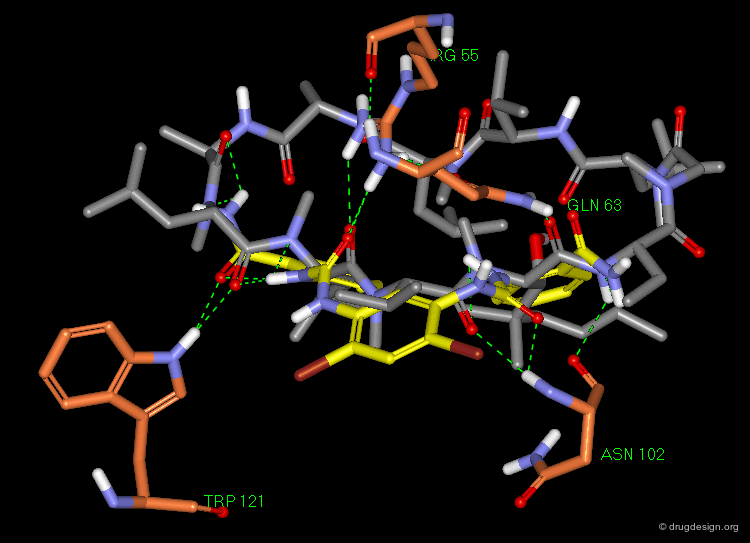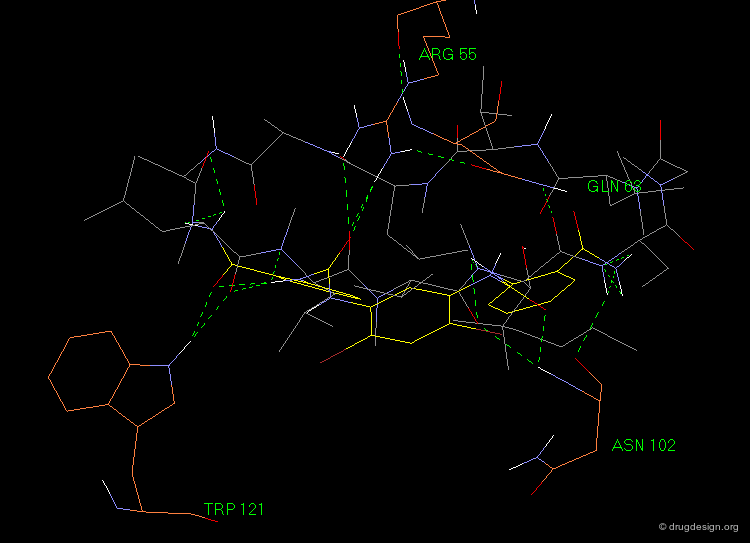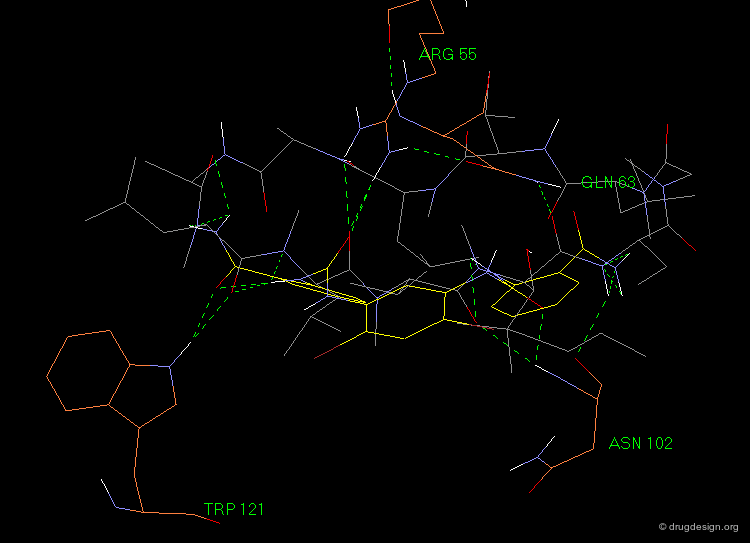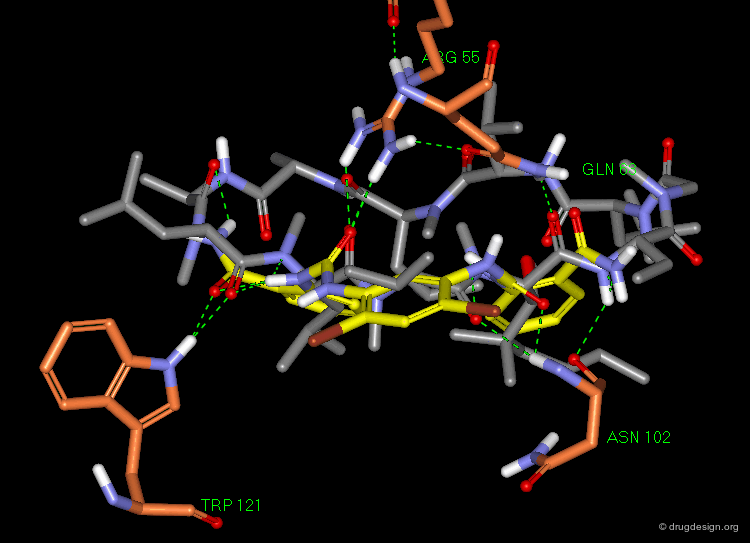
Optimization of Initial Hit¶
A combinatorial chemistry program was developed for generating SAR data. Analogs with improved biological activities were found, that were active at the sub-micromolar level.

Browser of Non-Peptidic Cyclophilin Ligands¶

Case Study-3 : Motilin Receptor Antagonists¶
Motilin Receptor Antagonists¶
Motilin is a 22 amino acid peptide secreted in the small intestine. This peptide hormone stimulates the contraction of smooth muscle in the gastrointestinal tract. Many motility disorders are currently treated through diet and stress management; an orally active small molecule motilin antagonist would be a useful agent for regulating gastrointestinal motility.

Motilin Receptor and Motilin Peptide¶
The motilin receptor belongs to the family of seven-transmembrane domain G-Protein Coupled Receptors (GPCRs). The difficulties in obtaining significant amounts of pure and active recombinant GPCRs have rendered the determination of high resolution X-ray structures very challenging. The structural analyses have been limited up to now to the study of the motilin peptide ligand alone.
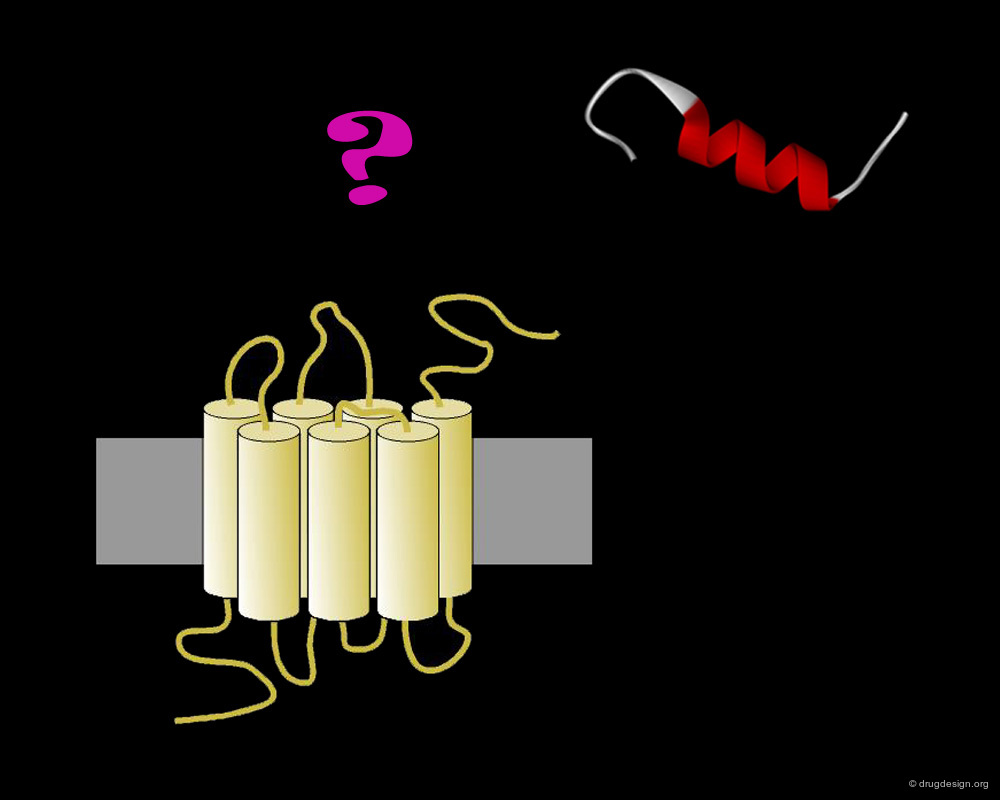
Structural Analyses on Motilin¶
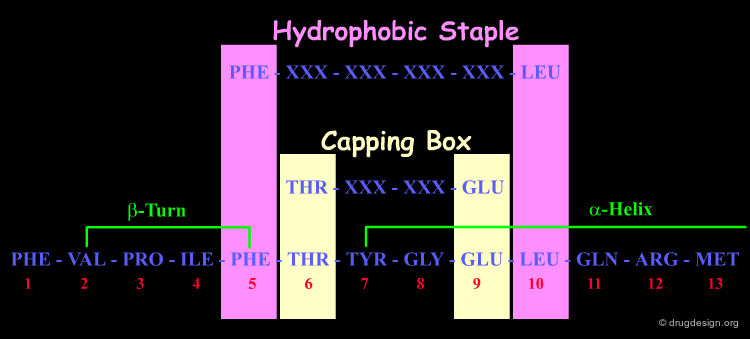
NMR analyses support the presence of a β-turn encompassing residues Val-2, Pro-3, Ile-4 and Phe-5.

Analyses of Motilin Folding¶
Amino-acid fingerprint matching indicates a potential for motilin to fold into both an N-terminal capping box (Thr-6 and Glu-9) and a hydrophobic staple motif (Phe-5 and Leu-10).
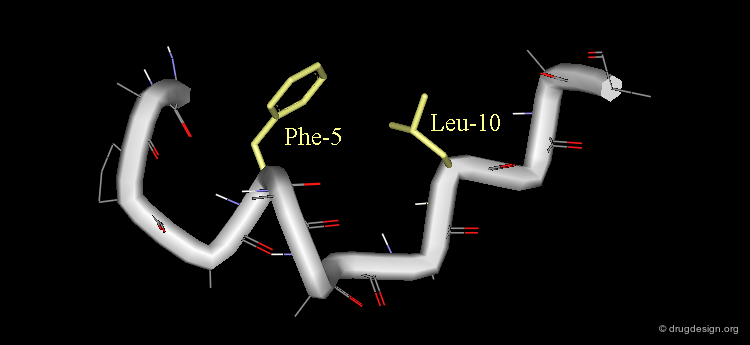
Bioactive Conformation of Motilin¶
The combination of the NMR and fingerprint analyses defines a motilin molecular geometry that can serve as a starting point for the identification of a 3D pharmacophore.
Biologically Relevant Residues of Motilin¶
Single point mutations and biological studies of the motilin mutants revealed that the most important amino-acid residues of the peptide hormone are the following: Phe-1, Ile-4 and Tyr-7.

The Motilin Pharmacophore¶
A pharmacophore model could be constructed, based on the 3D model of the bioactive conformation of motilin and Phe-1, Ile-4 and Tyr-7, the known biologically relevant residues.

RWJ-64583: a Trisubstituted Cyclopentene Lead¶
Sixty-three compounds were tested in a primary binding assay. They were selected from an initial list of 108 hits in the database search on the J&J corporate compound library. A promising chemical series emerged from the search, represented by an initial trisubstituted cyclopentene lead, RWJ-645833 with a Ki value of 461 nM.


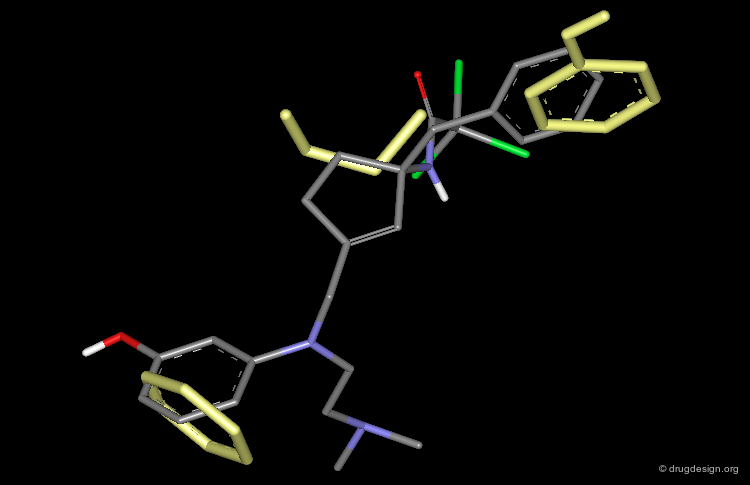
The Three-point Pharmacophore and RWJ-64583¶
The following view illustrates the superimposition of RWJ-64583 with the three-point pharmacophore query derived from motilin.
Optimization of the Initial Lead Molecule¶
The initial lead discovered was used as a starting point for the development of a directed medicinal chemistry synthetic program, resulting in the discovery of RWJ-67849 with a Ki value of 40 nM.


Mimicking the Phe-5 of Motilin¶
The extra urea phenyl group of RWJ-67849 was incorporated to mimic the Phe-5 side chain of motilin and thus allow for an additional binding moiety on the ligand. The two enantiomers of the racemic RWJ-67849 were resolved, and the R enantiomer (RWJ-68023) proved to be very active, in binding (Ki = 18 nM), in cell-based functional assays and other pharmacological tests.

Case Study-4 : Inhibitors of HIV-1 Protease¶
HIV-1 Protease Inhibition¶
Inhibition of the human immunodeficiency virus (HIV) protease has proven to be an effective concept for the development of drugs against the acquired immunodeficiency syndrome (AIDS). It has been shown that this enzyme is necessary for viral replication. The HIV-1 protease enzyme is a significant target protein in AIDS research.

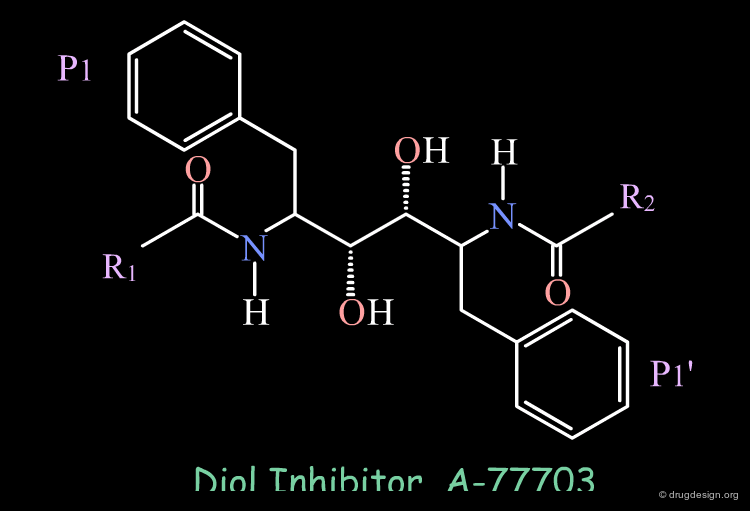
The Peptide Problem¶
Using the transition-state inhibitor approach, peptide inhibitors were conceived by replacing the scissile bond of the peptide substrate by a isostere that is not cleaved; A-77703 is an example of inhibitor containing the "dihydroxyethylene" moiety. Since these inhibitors showed low oral activity, new inhibitors with good bioavailablity were designed based on a peptidomimicry approach.
articles
Symmetry-based Inhibitors of HIV Protease. Structure-activity Studies of Acylated 2,4-Diamino-1,5-Diphenyl-3-Hydroxypentane and 2,5-Diamino-1,6-Diphenylhexane-3,4-Diol Kempf DJ, Codacovi L, Wang XC, Kohlbrenner WE, Wideburg NE, Saldivar A, Vasaanonda S, Marsh CK, Bryant P, Sham HL, Green BE, Betebenner DA, Erickson J and Norbeck DW J. Med. Chem. 36 1993
Database Searching for Non-Peptidic Scaffolds¶
Based on the docked diol/HIV-1 protease complex, a simple pharmacophore was defined and used as a query for 3D database searching.

articles
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
The Terphenyl Derivative Hit¶
The following molecule was identified by the database searching program and conformed well to the requirements of the input query.
articles
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
Analysis of the Content of the Hit¶
Docking experiments of the terphenyl derivative in the HIV-1 protease showed that the top methoxy group was mimicking the structural water and the bottom hydroxy and methoxy groups bind to the Asp-25.

articles
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
Replacing Cyclohexanone by a 7-Membered Ring¶
Modeling studies were performed and showed that replacing the cyclohexanone by a cyclo-urea (7 membered ring) increased the binding to the enzyme. It can be seen that this change provides a better hydrogen bonding to the Asp-25 and Asp-25'

Problem of the 6-membered Ring¶
The problem with the six-membered ring hit is that it doesn't have the proper substitution pattern to allow access to the two other hydrophobic pockets.

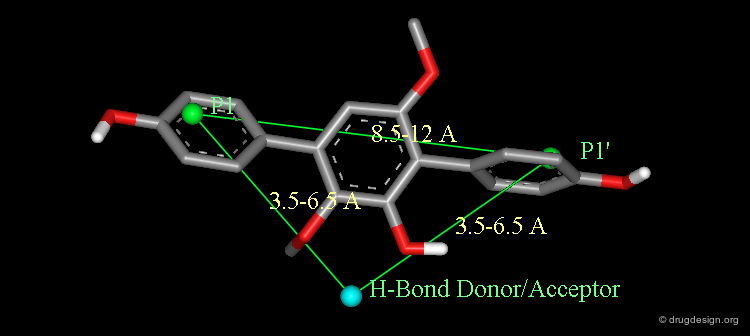
Optimal Framework: 7-membered Cyclic Urea¶
The seven-membered cyclic urea ring provides an optimal framework both in terms of conformation and stereochemistry. The substitution pattern of the central scaffold allows the access to the four hydrophobic pockets of the enzyme corresponding to P1, P1', P2 and P2'.

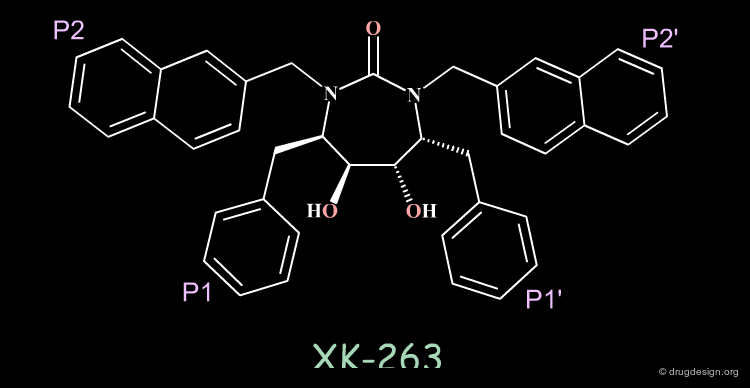
Design of Cyclic Urea Scaffold¶
All this design and modeling led to the highly potent cyclic urea XK 263 (pKi = 0.31nM).
XK-263 is a Non-Peptidic Mimic of A-77003¶
The alignment of the non-peptidic XK-263 (white) on the peptidic A-77003 (blue) illustrates how a non-peptidic molecule can imitate the essential 3D features of a reference peptide compound; both compounds are potent inhibitors.
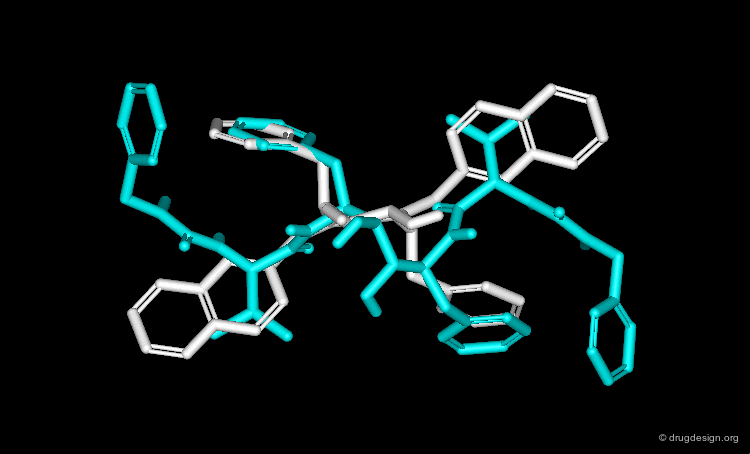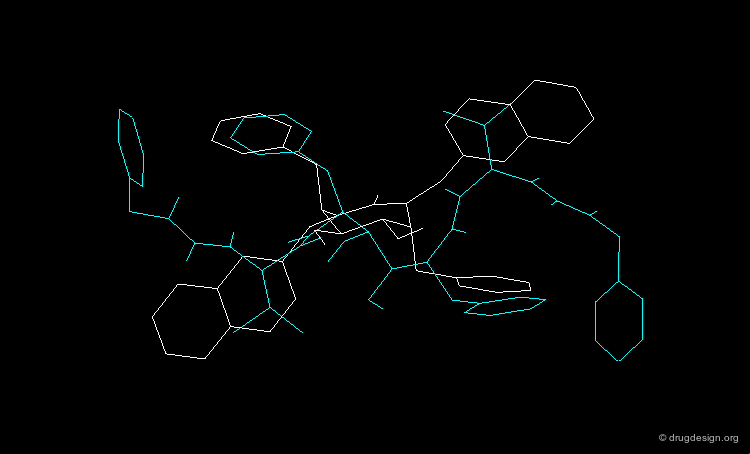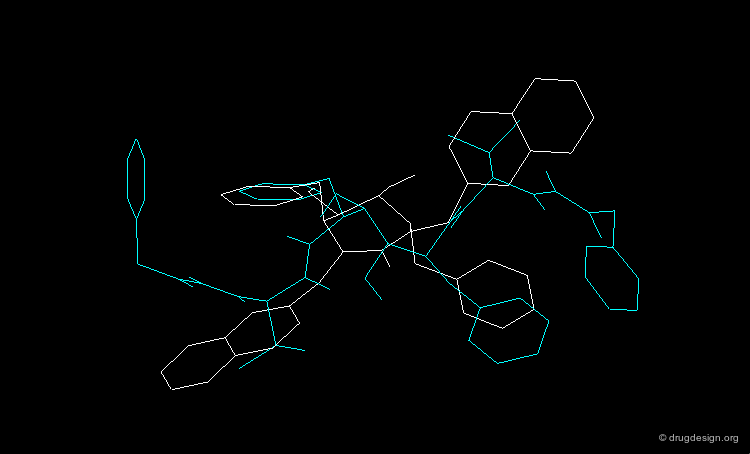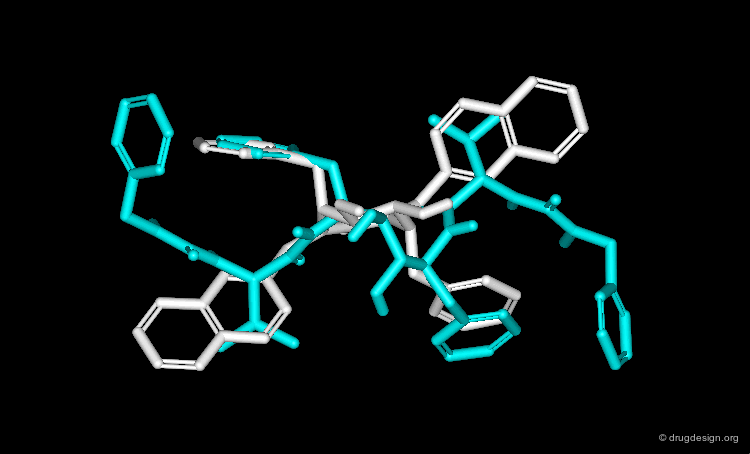
articles
Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang C-H, Weber PC, Jacson DA, Sharpe TR and Erickson-Viitanen S Science 263 1994
Summary¶
In this example 3D database searching permitted the emergence of an idea (the consideration that a cyclic scaffold can mimic both the peptide backbone and the structural water). This idea was further exploited by combining structure-based drug design and chemical creativity (see scheme below). 3D database searching is a method of choice for generating new ideas, with the advantage that 'hits' do not need to be available in the stockroom.

Case Study-5 : Non-Sugar Antagonists of Selectin¶
Reference Compound¶
Antagonists of selectins are of interest in stopping the progression of the clinical manifestations of inflammatory disease. The sialyl-Lewis-X (sLex) binds to these proteins and can serve as a basis for the design of antagonists. As the development of a carbohydrate structure is not easy, a 3D pharmacophore-based approach was considered for discovering simple non-sugar mimics.
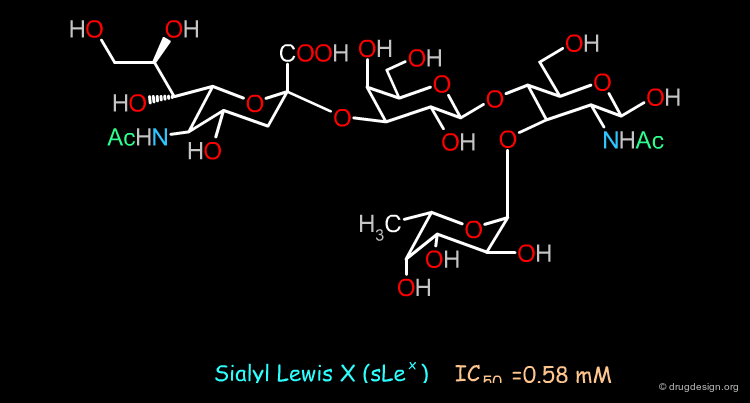
Initial SAR Analyses¶
Preliminary SAR studies revealed that the sulfo-Lex derivative 1 was a selectin blocker. Moreover further SAR indicated that the lactose moiety of compound 1 could be replaced by a Ser-Glu dipeptide (compound 2).

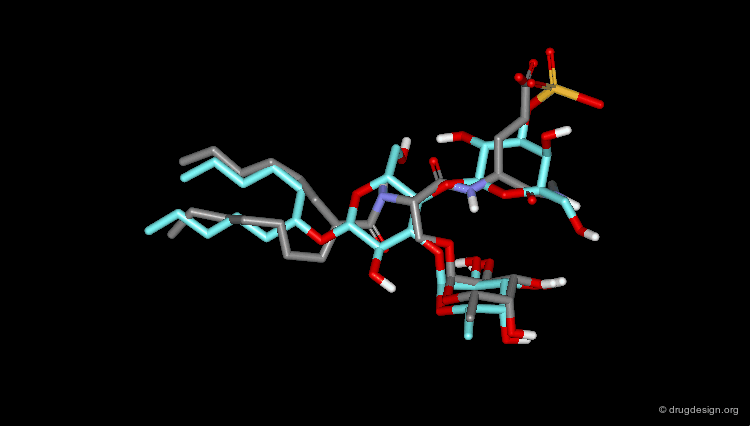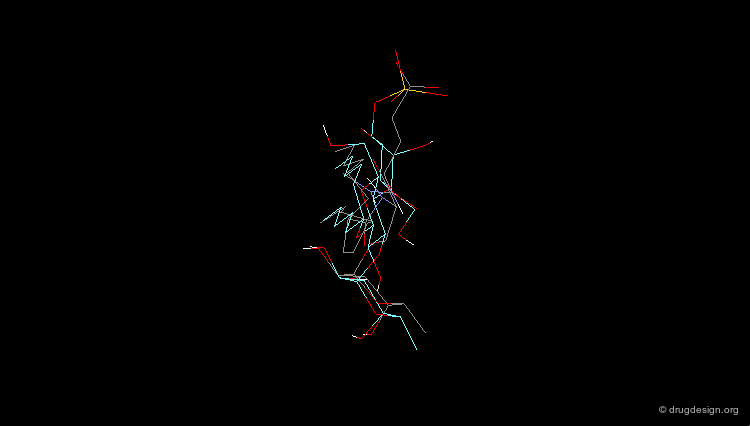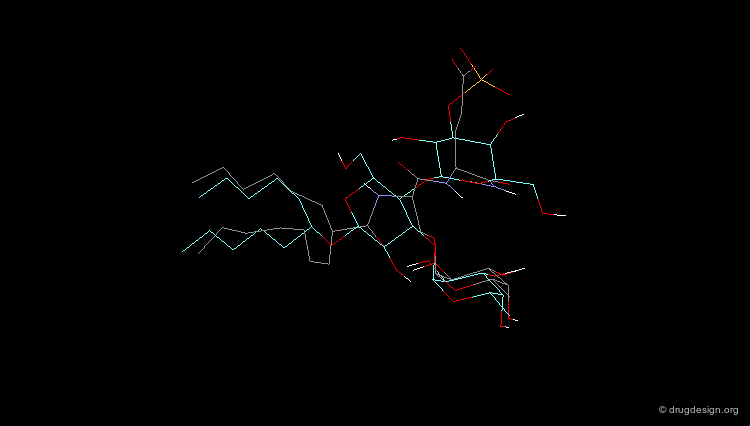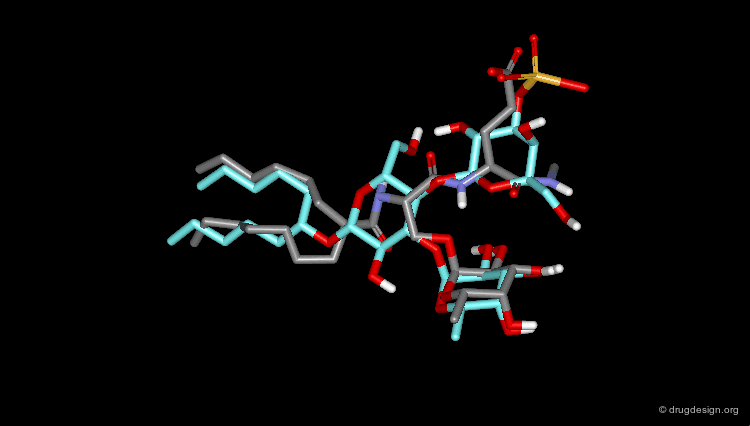
Pharmacophore Model¶
A 3D pharmacophore was derived that exploited the information from the SAR. Based on an initial pharmacophore shown on the left side, no hits were obtained. The model was therefore modified by replacing (1) the fucose unit by a carboxylic acid (for calcium binding); (2) the negatively charged sulfonic group was replaced by a carboxylic acid and (3) the branched alkyl chain was replaced by a single aliphatic chain.


Results of 3D Searching¶
The modified pharmacophore yielded 3, a molecule that showed inhibitory activity against E-selectin. Despite its inferior biological activity as compared to the reference compounds, this hit was considered of sufficient interest for further improvements, because of its non-sugar structure.

Optimization of the Diphenyl Ether Hit¶
Aligning the 3D structures of 1 and 3 revealed an unfavorable repulsion in the hit between the carboxylic acid of the benzoic acid moiety and a carboxylic acid of the isophtalic fragment. In addition, based on the X-ray structure of E-Selectin, modeling analyses indicated that one carboxylic acid of the isophtalic acid moiety did not contribute to the binding.

Optimization of the Diphenyl Ether Hit¶
These observations suggested that one carboxylic acid of the isophthalic acid moiety might be removed. As the decarboxy derivative of 3 could not be synthesized, the related analog 4 was prepared with the intention of testing this hypothesis in this series. Compound 4 proved to have a very weak biological activity.

Optimization of the Diphenyl Ether Hit¶
However the decarboxy derivative 5 was found with good antagonistic activity. This observation is consistent with the idea of low energy of the bioactive conformation of the molecule when it binds to E-selectin.

Summary¶
This study illustrates that non-sugar antagonists of the E-selectin protein can be discovered using a 3D database searching approach. Starting with sugar-based derivatives like 1 and 2, the 3D searching led to the discovery of the non-sugar mimetic antagonist 5.

Browser of Selectin Antagonists¶

Case Study-6 : Dopamine Transporter Inhibitors¶
The Dopamine Transporter Target¶
The work presented in this topic was carried out by the group at the University of Michigan to find a replacement therapy for cocaine addiction. They took the dopamine transporter (DAT) as a molecular target after obtaining biological results showing that cocaine binds to DAT, and other data suggesting that drugs that bind to biogenic amine transporters, including DAT, might be effective in combating cocaine addiction.
Methodology: 3D Database Searching¶
A 3D database approach was used in which successive pharmacophore models were utilized and refined. The search was based on either the Available Chemical Directory (ACD), or the National Cancer Institute (NCI) 3D databases containing readily available molecules that can be biologically tested quickly. We start by presenting the first pharmacophore model.

First Pharmacophore¶
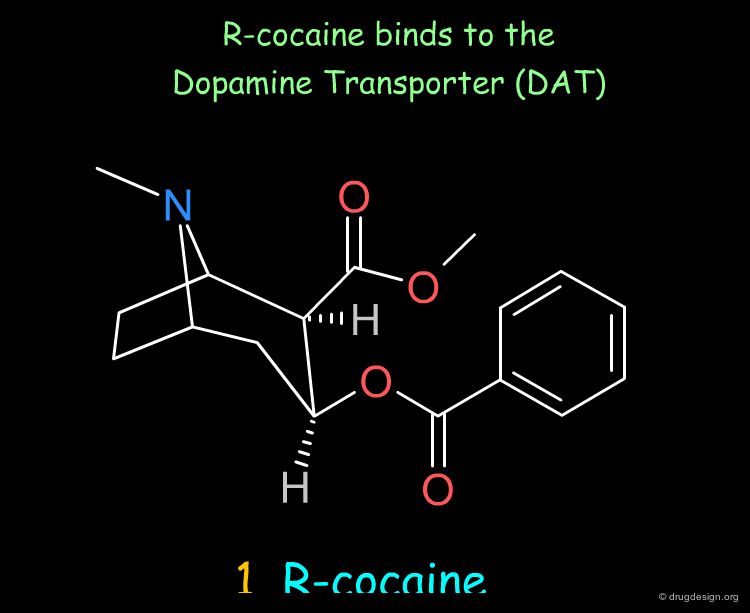
The first pharmacophore model was directly derived from the 3D structure of cocaine, and assumed that the following groups were important: the tertiary amine (of the tropane), the phenyl (of the phenyl ester) and the carbonyl (of the methyl ester), at distances d1, d2 and d3 with some tolerance, as indicated below.

articles
Discovery of a novel dopamine transporter inhibitor, 4-hydroxy-1-methyl-4-(4-methylphenyl)-3-piperidyl 4-methylphenyl ketone, as a potential cocaine antagonist through 3D-database pharmacophore searching. Molecular modeling, structure-activity relationships, and behavioral pharmacological studies. Wang S, Sakamuri S, Enyedy IJ, Kozikowski AP, Deschaux O, Bandyopadhyay BC, Tella SR, Zaman WA, Johnson KM. J Med Chem. 43(3) 2000
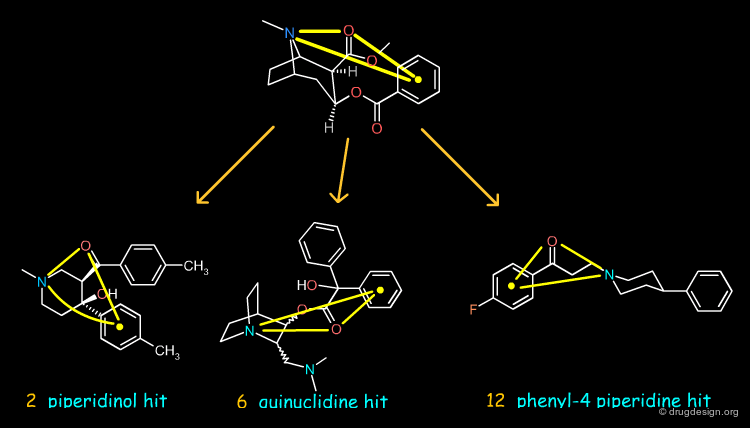
3D Searching Results with the First Pharmacophore¶
Searching in the NCI 3-D database of "open" compounds led to the identification of molecules in line with the first hypothesis. The piperidinol (2), the quinuclidine (6) and the phenyl-4 piperidine (12) compounds proved to inhibit the activity of the DAT protein at the micromolar range. Below, we describe the attempts made by the group at the University of Michigan for each of these to obtain more potent compounds.
articles
Discovery of a novel dopamine transporter inhibitor, 4-hydroxy-1-methyl-4-(4-methylphenyl)-3- piperidyl 4-methylphenyl ketone, as a potential cocaine antagonist through 3D-database pharmacophore searching. Molecular modeling, structure-activity relationships, and behavioral pharmacological studies. Wang S, Sakamuri S, Enyedy IJ, Kozikowski AP, Deschaux O, Bandyopadhyay BC, Tella SR, Zaman WA, Johnson KM. J Med Chem. 43(3) 2000
Piperidinol Hit¶
Let us begin with the piperidinol hit. This compound proved to be a DAT inhibitor with a fairly good biological activity, as indicated by an inhibition constant Ki of 0.360 µM (note that R-cocaine has a Ki of 0.27 µM).

articles
Discovery of a novel dopamine transporter inhibitor, 4-hydroxy-1-methyl-4-(4-methylphenyl)-3- piperidyl 4-methylphenyl ketone, as a potential cocaine antagonist through 3D-database pharmacophore searching. Molecular modeling, structure-activity relationships, and behavioral pharmacological studies. Wang S, Sakamuri S, Enyedy IJ, Kozikowski AP, Deschaux O, Bandyopadhyay BC, Tella SR, Zaman WA, Johnson KM. J Med Chem. 43(3) 2000
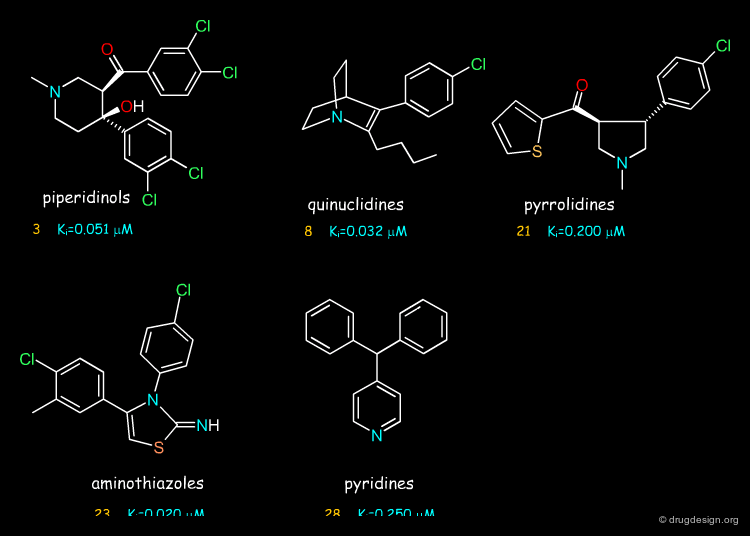
Optimization of the Piperidinol Hit¶
The hit was further explored. Compounds such as 3, 4 and 5 are examples of analogs that were synthesized, tested, and showed increased inhibitory activities.

articles
Pharmacophore-based discovery, synthesis, and biological evaluation of 4-phenyl-1-arylalkyl piperidines as dopamine transporter inhibitors. Sakamuri S, Enyedy IJ, Kozikowski AP, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 11(4) 2001
Quinuclidine Hit¶
The second hit, with a quinuclidine nucleus, was ordered and tested biologically; it inhibited DAT with a Ki of 8.9 µM.

Optimization of the Quinuclidine Hit¶
Optimization of the quinuclidine hit led to the discovery of more potent analogs. In particular, molecules such as 7, 8 and 9 are examples of compounds that had improved inhibitory potencies.

articles
Synthesis of 2-alkyl-3-aryl-substituted quinuclidines as novel dopamine transporter inhibitors. Sukumar Sakamuri, Istvan J. Enyedy, Alan P. Kozikowski and Shaomeng Wang. Tetrahedron letters. 41 2000
2,3-Disubstituted quinuclidines as a novel class of dopamine transporter inhibitors. Sakamuri S, Enyedy IJ, Zaman WA, Tella SR, Kozikowski AP, Flippen-Anderson JL, Farkas T, Johnson KM, Wang S. Bioorg Med Chem. 11(6) 2003
Phenyl-4 Piperidine Hit¶
The third hit 12 (with a phenyl-4 piperidine moiety) proved to be a DAT inhibitor with a Ki of 1.56 µM.

articles
Pharmacophore-based discovery, synthesis, and biological evaluation of 4-phenyl-1-arylalkyl piperidines as dopamine transporter inhibitors. Sakamuri S, Enyedy IJ, Kozikowski AP, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 11(4) 2001
Optimization of the Phenyl-4 Piperidine Hit¶
The phenyl-4 piperidine derivative 12 was further optimized. Compound 13 is an example of analog that was found to have an improved inhibition constant (Ki = 0.09 µM).

articles
Pharmacophore-based discovery, synthesis, and biological evaluation of 4-phenyl-1-arylalkyl piperidines as dopamine transporter inhibitors. Sakamuri S, Enyedy IJ, Kozikowski AP, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 11(4) 2001
Challenging the First Pharmacophore¶
Hits were found with the first pharmacophore model however, their optimization failed to result in nanomolar activities. Using the fact that very potent known DAT inhibitors such as 12, 13, 14, 15 do not have a carbonyl fragment, the Michigan group tried to assess the validity of the pharmacophore hypothesis, which includes a carbonyl moiety.

articles
2D QSAR modeling and preliminary database searching for dopamine transporter inhibitors using genetic algorithm variable selection of Molconn Z descriptors. Hoffman BT, Kopajtic T, Katz JL, Newman AH. J Med Chem. 43(22) 2000
Chemistry and Biology of the 2.beta.-Alkyl-3.beta.-phenyl Analogs of Cocaine: Subnanomolar Affinity Ligands That Suggest a New Pharmacophore Model at the C-2 Position. Alan P. Kozikowski, M. K. Eddine Saiah, Kenneth M. Johnson, and John S. Bergmann. J Med Chem. 38 1995
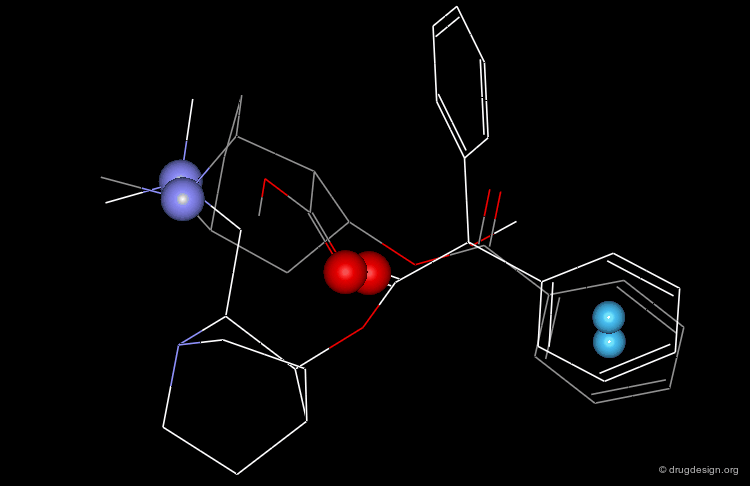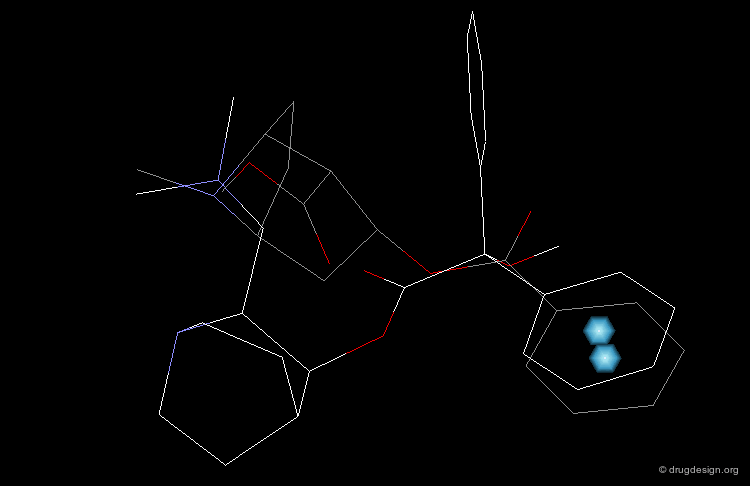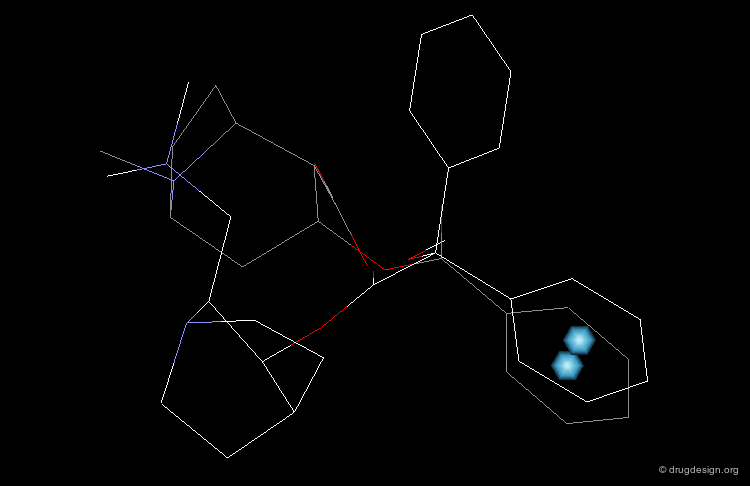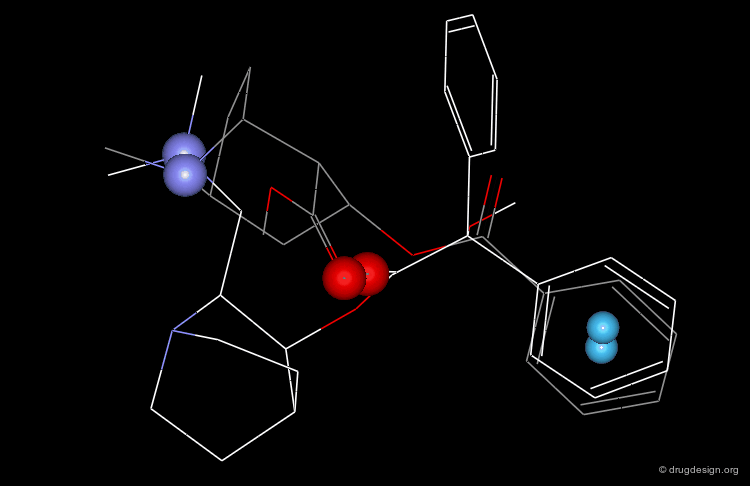
Structural Analyses of the Quinuclidine Hit¶
In particular, the Michigan group tried to identify which of the two nitrogen atoms of the quiniclidine hit mimics the one in cocaine. Modeling analyses indicated that the two hypotheses could be envisaged, but the 3D views revealed that in both cases, the remaining fragments of the two molecules did not overlap well (Sup1 and Sup2).


articles
2,3-Disubstituted quinuclidines as a novel class of dopamine transporter inhibitors. Sakamuri S, Enyedy IJ, Zaman WA, Tella SR, Kozikowski AP, Flippen-Anderson JL, Farkas T, Johnson KM, Wang S. Bioorg Med Chem. 11(6) 2003
Modeling Analyses: C=O Not Necessary!¶
Further modeling analyses revealed that a better superposition could be obtained when the carbonyl of the quinuclidine molecule is not viewed as a pharmacophoric moiety: rather, the cocaine carbonyl group can be mimicked by a simple alkyl chain.

Browser Associated to the First Pharmacophore¶
The different molecules examined for the first pharmacophore are visualized in the following browser.
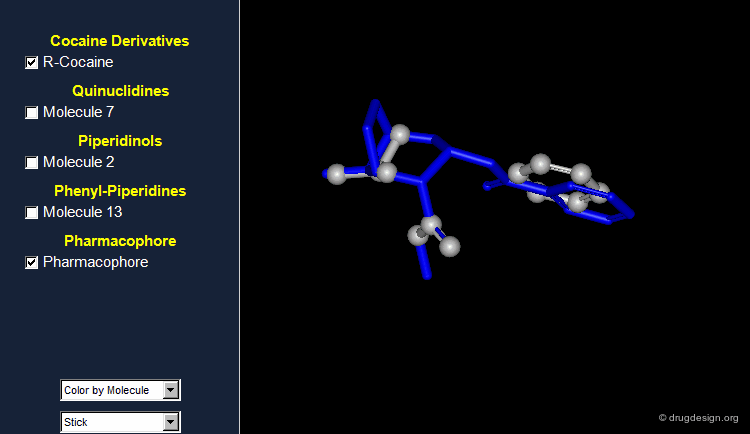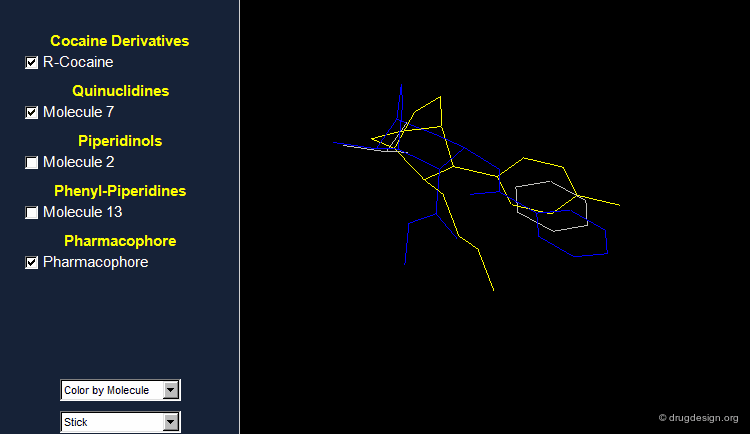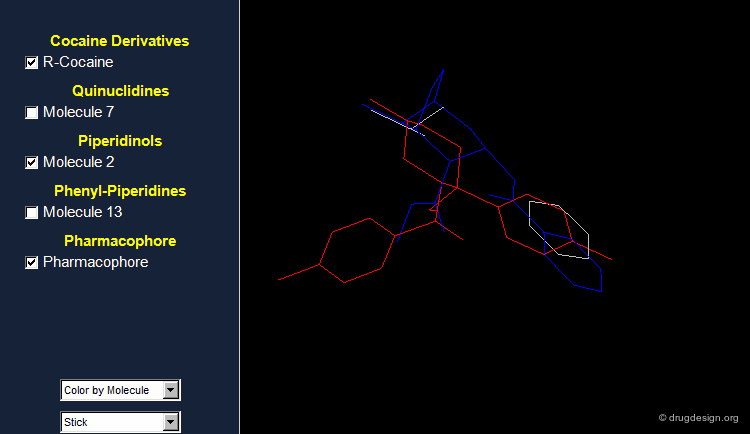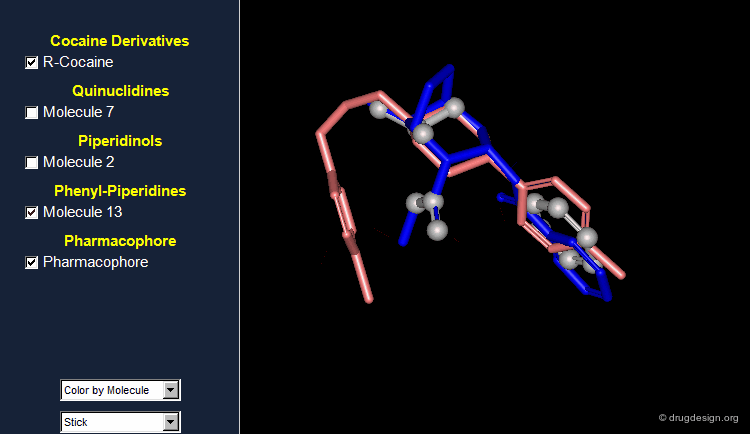
What Can Be Learned So Far?¶
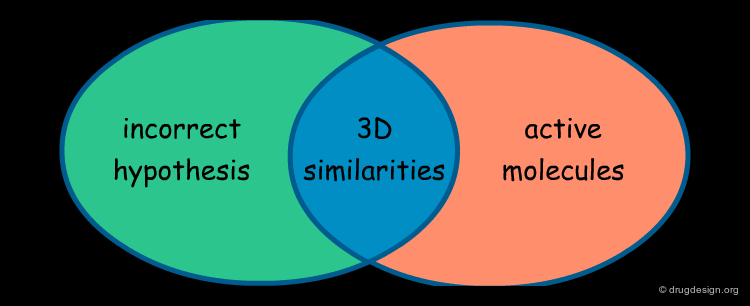
The first part of this story illustrates that: (1) it is useful to challenge a current hypothesis; (2) an incorrect 3D pharmacophore hypothesis can possibly lead to useful results. This is shown by the 3D searching process, which generates molecules that have certain 3D similarities with the reference active molecule from which the pharmacophore was derived.
Second Pharmacophore¶
The Michigan group continued to search for novel DAT inhibitors. Instead of using the cocaine molecule as a reference, they used the structure of the piperidinol compound 3 they had already discovered. Its 3D structure was therefore used to define a second 3D pharmacophore model.
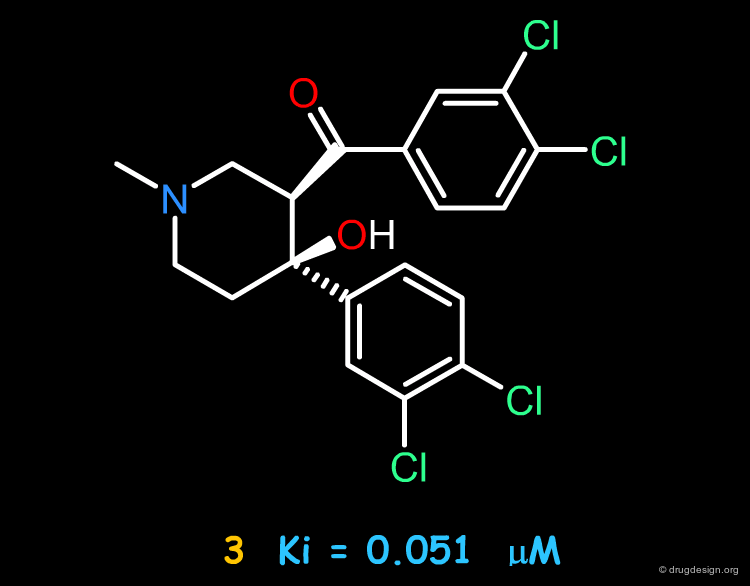
Characteristics of the Second Pharmacophore¶
The second pharmacophore consisted of a sp3 nitrogen atom and two phenyl rings at distances d1, d2 and d3, as measured in the structure of the piperidinol analog 3.
articles
Pharmacophore-based discovery of 3,4-disubstituted pyrrolidines as a novel class of monoamine transporter inhibitors. Enyedy IJ, Zaman WA, Sakamuri S, Kozikowski AP, Johnson KM, Wang S. Bioorg Med Chem Lett. 11(9) 2001
3D Searching with the Second Pharmacophore¶
3D searching with the second pharmacophore and the ACD chemical database yielded the pyrrolidine hit 17. The compound was ordered and proved to be a DAT inhibitor with a Ki of 1.4 µM.
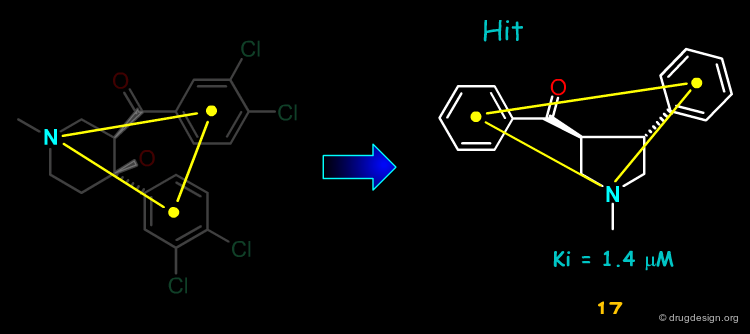
articles
Pharmacophore-based discovery of 3,4-disubstituted pyrrolidines as a novel class of monoamine transporter inhibitors. Enyedy IJ, Zaman WA, Sakamuri S, Kozikowski AP, Johnson KM, Wang S. Bioorg Med Chem Lett. 11(9) 2001
Optimization of the Pyrrolidine Hit¶
It was also found that many analogs of the pyrrolidine hit were commercially available, which permitted the optimization of 17 by ordering and testing them. The SAR analyses revealed the existence of more potent analogs such as 18, 19 and 20.

articles
Pharmacophore-based discovery of 3,4-disubstituted pyrrolidines as a novel class of monoamine transporter inhibitors. Enyedy IJ, Zaman WA, Sakamuri S, Kozikowski AP, Johnson KM, Wang S. Bioorg Med Chem Lett. 11(9) 2001
What Can Be Learned So Far?¶
It seems only logical to ask why the Michigan group used the piperidinol inhibitor, since there are DAT inhibitors that are a hundred times more potent. The real reason is that they wanted to exploit their piperidinol hit to find another scaffold that could be easily optimized. This strategy failed, however: the optimized compounds were not potent enough.
Browser Associated to the Second Pharmacophore¶
The different molecules examined for the second pharmacophore are visualized in the following browser.

Third Pharmacophore¶
Because they needed more potent compounds, the Michigan group considered a third pharmacophore model, based on the structure of a drug currently used for the treatment of opioid-induced sedation: Mazindol (note that this compound represents another class of inhibitors that has a different mode of interaction with DAT, as compared to cocaine).

articles
Dopamine transporter site-directed mutations differentially alter substrate transport and cocaine binding. Kitayama S, Shimada S, Xu H, Markham L, Donovan DM, Uhl GR. Proc Natl Acad Sci U S A. 89(16) 1992
Dopamine Transporter Transmembrane Domain Polar Mutants: G and G Values Implicate Regions Important for Transporter Functions. Masanari Itokawa, Zhicheng Lin, Ning-Sheng Cai, Cindy Wu, Shigeo Kitayama, Jia-Bei Wang, and George R. Uhl. Mol Pharmacol. 57 2000
2-Carbomethoxy-3-aryl-8-oxabicyclo[3.2.1]octanes: potent non-nitrogen inhibitors of monoamine transporters. Meltzer PC, Liang AY, Blundell P, Gonzalez MD, Chen Z, George C, Madras BK. J Med Chem. 40(17) 1997
Halogenated Mazindol Analogs as Potential Inhibitors of the Cocaine Binding Site at the Dopamine Transporter. William J. Houlihan, John W. Boja, Vincent A. Parrino, Theresa A. Kopajtic, and Michael J. Kuhar. J Med Chem. 39(25) 1996
WIN 35,428 and mazindol are mutually exclusive in binding to the cloned human dopamine transporter. Xu C, Reith ME. J Pharmacol Exp Ther. 282(2) 1997
Discovery of substituted 3,4-diphenyl-thiazoles as a novel class of monoamine transporter inhibitors through 3-D pharmacophore search using a new pharmacophore model derived from mazindol. Enyedy IJ, Wang J, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 12(13) 2002
Bioactive Form of Mazindol¶
Mazindol can exist in two tautomeric forms: a carbinol form A, and a keto form B. As the biological action occurs at a pH slightly above 7, the predominant tautomer of this compound is represented by the tricyclic carbinolamine form A.

articles
Discovery of substituted 3,4-diphenyl-thiazoles as a novel class of monoamine transporter inhibitors through 3-D pharmacophore search using a new pharmacophore model derived from mazindol. Enyedy IJ, Wang J, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 12(13) 2002
Characteristics of the Third Pharmacophore¶
The third pharmacophore was derived from the structure of mazindol and consisted of a sp2 nitrogen atom and two phenyl rings at distances d1, d2 and d3, as found in the carbinolamine tautomer of mazindol.
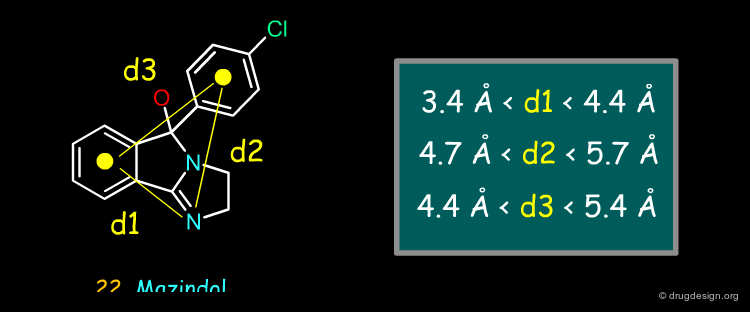
articles
Discovery of substituted 3,4-diphenyl-thiazoles as a novel class of monoamine transporter inhibitors through 3-D pharmacophore search using a new pharmacophore model derived from mazindol. Enyedy IJ, Wang J, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 12(13) 2002
3D Searching with Third Pharmacophore¶
3D searching with the third pharmacophore and the ACD chemical database yielded a whole family of aminothiazole analogs as candidate hits (the database contained many such analogs). Subsequent ordering and testing revealed that these compounds represented a novel series of DAT inhibitors, as exemplified by compounds 23, 24, 25 and 26.

articles
Discovery of substituted 3,4-diphenyl-thiazoles as a novel class of monoamine transporter inhibitors through 3-D pharmacophore search using a new pharmacophore model derived from mazindol. Enyedy IJ, Wang J, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 12(13) 2002
Browser Associated to the Third Pharmacophore¶
The different molecules examined for the third pharmacophore are visualized in the following browser.

Fourth Pharmacophore¶
In an attempt to discover lead compounds with significantly improved potencies, the Michigan group finally considered a fourth pharmacophore model based on known potent DAT inhibitors of other groups such as 14, 15, 16, 17 and also their own piperidinol lead 2.

Aligning Low Energy Conformers¶
Modeling analyses were carried out to identify common structural features of the different and chemically unrelated potent analogs under consideration. Low energy conformers of the different structures were aligned, which enabled the characterization of the fourth pharmacophore.

articles
Pharmacophore-based discovery of substituted pyridines as novel dopamine transporter inhibitors. Enyedy IJ, Sakamuri S, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 13(3) 2003
Characteristics of the Fourth Pharmacophore¶
The fourth pharmacophore was thus derived from the alignment of low energy conformers of the potent compounds previously described and consisted of a sp3 nitrogen atom and two phenyl rings at distances d1, d2 and d3, as measured in the superimposition.


articles
Pharmacophore-based discovery of substituted pyridines as novel dopamine transporter inhibitors. Enyedy IJ, Sakamuri S, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 13(3) 2003
3D Searching with the Fourth Pharmacophore¶
Substituted pyridine derivatives emerged as hits by searching in the NCI 3-D database of "open" compounds. Molecules such as 27 and 28 proved to possess DAT inhibitory activities; they represented a rather simple class of new DAT inhibitors.

articles
Pharmacophore-based discovery of substituted pyridines as novel dopamine transporter inhibitors. Enyedy IJ, Sakamuri S, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 13(3) 2003
Optimization of the Substituted Pyridine Hit¶
Several pyridine analogs were ordered and tested to explore the structure-activity relationships of this new class of DAT inhibitors. They contained a pyridine and two phenyl rings connected differently, and their corresponding biological activities are described below.

articles
Pharmacophore-based discovery of substituted pyridines as novel dopamine transporter inhibitors. Enyedy IJ, Sakamuri S, Zaman WA, Johnson KM, Wang S. Bioorg Med Chem Lett. 13(3) 2003
Browser Associated to the Fourth Pharmacophore¶
The different molecules examined for the fourth pharmacophore are visualized in the following browser.
Summary¶
This study illustrates that it may be advantageous to generate many hypotheses, then refine them, and integrate results derived from other sources. This methodology led to the discovery of six novel classes of DAT inhibitors, based on rather simple pharmacophore models.

ADDITIONAL CASE STUDIES¶
Additional Case Studies¶

Copyright © 2024 drugdesign.org