Structural Bioinformatics¶
Info
Structural Bioinformatics is a discipline that unites experimental and theoretical fields that all have the 3D structures of macromolecules in common. It aims at understanding how biological systems work at the molecular level. In this chapter we present the organization of macromolecules, their 3D determination, visualization and structural classification together with the principles of molecular recognition, structure-based drug design, and applications of physical and chemical principles with algorithms for the simulation of macromolecular structures.
Number of Pages: 135 (±3 hours read)
Last Modified: June 2008
Prerequisites: None
Introduction to Structural Bioinformatics¶
Challenges in the Post Genomic Era¶
The completion of the human genome project, which successfully determined the sequence of human DNA, was the first step towards a comprehensive understanding of life at the molecular level. This heralded the dawn of a new era and assigned researchers and scientists the challenge of analyzing, deciphering and understanding the genetic origin of human disease. One of the key issues is the characterization and function of proteins with a view to using the findings in the discovery of novel and effective therapeutic treatments.

The Informational Chaos¶
As a consequence of advances in the fields of molecular biology, genomics, proteomics, structural biology and drug discovery, the post-genomic era has been characterized by the uncontrolled explosion of information. This includes vast amounts of genetic data, identification of numerous potential targets, large amounts of biological information, huge amounts of solved crystal structures, large quantities of SAR data from HTS, etc. New strategies are absolutely crucial if we are to face the challenges of the post-genomic era.

Integration through Computational Science¶
To be able to handle the vast volumes of data generated by new high-throughput technologies, new high-throughput computational disciplines have emerged to face this challenge: computational biology to solve problems in biology; bioinformatics to solve problems in genomics; computational chemistry to solve problems in chemistry and cheminformatics to solve problems in drug discovery.

Structural Bioinformatics¶
Structural Bioinformatics is an interdisciplinary field that deals with the three dimensional structures of biomolecules. It attempts to model and discover the basic principles underlying biological machinery at the molecular level. It is based on the belief that 3D structural information of a biological system is the core to understanding its mechanism of action and function. Structural bioinformatics combines applications of physical and chemical principles with algorithms from computational science.

wikipedia
Grouping Fields into One Discipline¶
For the lay person, it can be confusing to figure out where structural bioinformatics fits into other disciplines. Although it emerged as a new discipline in the 2000s, it was in fact born in the early 1950s as a result of the crystallographic determinations of hemoglobin and myoglobin. Structural bioinformatics has now become an interdisciplinary field with scientists that are all involved in the 3D structures of macromolecules.

wikipedia
3D Basis of Structural Bioinformatics¶
Structural Bioinformatics is based on the protein structure/function paradigm, which states that the function of a macromolecule is dictated by its 3D-structure. As a consequence, if we want to predict the function of a protein we need to start from its 3D representation and try to decode the hidden information embedded in this structure. The challenges for our post genomic era towards a systematic understanding of life are: (1) to obtain the 3D structures of all the proteins encoded in the genomes and (2) to decode their function based on their structure.

The Structural Genomics Effort¶
"Structural Genomics" aims at obtaining the 3D structures of all the proteins encoded in the genome in a systematic way. Since protein folding remains an unsolved problem, theoretical approaches to translating sequences into 3D protein structures are unfeasible; and scientists have to rely mainly on experimental determinations. A number of worldwide structural genomics programs are in progress; their common aim is to make large-scale, high-throughput protein structure determinations.

articles
Making decisions for structural genomics Rodrigues A, Hubbard R.E Briefings in Bioinformatics 4 2003 10.1093/bib/4.2.150
wikipedia
The Protein Structure Initiative¶
The National Institute of General Medical Sciences (NIGMS/NIH) founded in 2000 one of the first structural genomics programs: the "Protein Structure Initiative" (PSI). The goal of the project is to reveal the exact 3D structure of all proteins of the genomes by experimental methods or computational approaches. The PSI has started to create a large collection of protein structures determined by high-throughput techniques using X-ray crystallography and NMR spectroscopy.

wikipedia
Strategy of the Protein Structure Initiative¶
Structure determination is a long and expensive process. To achieve its goal the NIGMS classifies proteins into families and concentrates its effort on experimentally solving the 3D structure of only one member of each family. All the other members of a family can be modeled using homology modeling techniques.

wikipedia
The Structural Genomics Consortium¶
Due to the formidable challenges posed by the structural genomics effort, other pragmatic initiatives were taken based on a prioritization of the targets to be achieved. For example, the "Structural Genomics Consortium" was created in 2004 with the aim of solving the three dimensional structures of proteins known to be of medical relevance. The Consortium has defined a list of 2000 disease-relevant proteins. In 4 years of activities the consortium has solved 386 proteins and plans to solve 50% of the proteins on the list by 2011.

Global Planning of Structural Genomics¶
The key difference between structural genomics and traditional structural biology lies in the global strategy adopted by structural genomics to cover the entire area of protein structures. The strategy consists of selecting targets with high informational content, avoiding redundancy and systematically spanning the entire molecular space. This global planning has paved the way to the determination of 3D structures of all the genome proteins.


The Impact of Structural Genomics¶
About 20% of the new structures deposited in the PDB are from SG, but the real contribution of SG is in new structures from families that do not have 3D representatives. In 2005 only 36% of the Pfam families (2736 of 7677) had a 3D representative. In their study, Chandonia and Brenner analyzed the evolution of the contribution of SG, and showed that in 2005 about 50% of the total number of first representatives of protein families had been solved by SG organizations.

articles
The impact of structural genomics: expectations and outcomes. Chandonia, J. M. and Brenner, S. E Science 311 2006
wikipedia
The Relationship between Structure and Function¶
The comparison between sequence similarities and the 3D structures of proteins has become a major source of discoveries and predictions in biology. Much of the information deduced from the structure may provide important clues to guide researchers towards function. Many properties that are not apparent in the sequence (e.g. spatial proximity of amino acids) are apparent in the 3D structure.
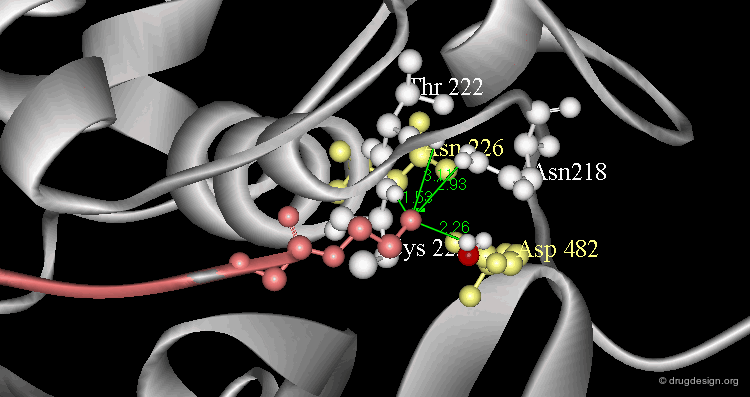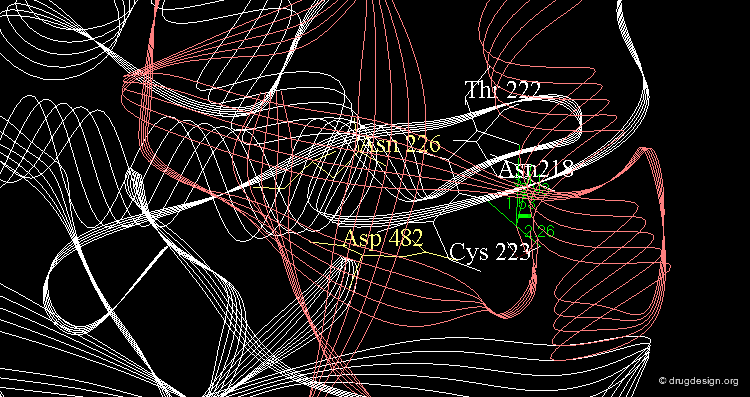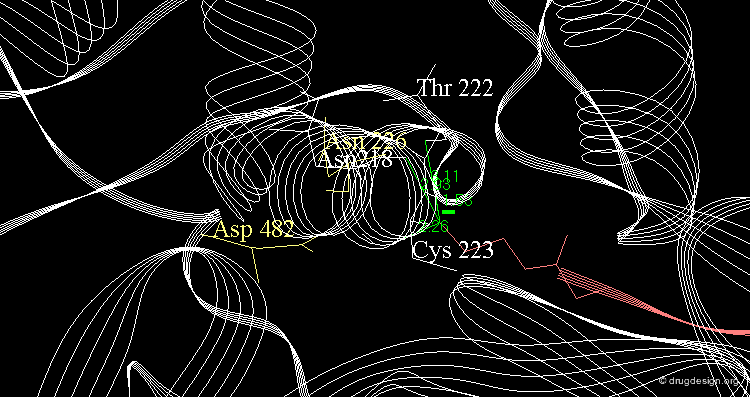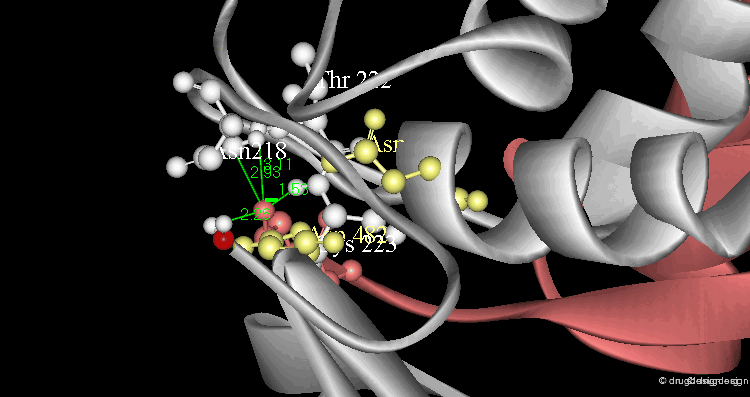
Example of a Structure-Function Relationship¶
Subtilisin and chymotrypsin are two proteins that take on completely different folds despite the fact that the two proteins have identical functions: they break peptide bonds. Their common function is due to the presence of three residues (Asp, His, Ser) forming a catalytic triad that has exactly the same geometric arrangement, although located at different positions in the primary sequence.


book
Petsko G.A. and Ringe D. Protein Structure and Function Sinauer Associates 2004
wikipedia
Learning from Evolution¶
Evolution provides an important key to function by taking advantage of the similarity principle to generate information by comparative analyses. Typically, homolog proteins (descended from a common ancestor) have significant sequence similarity and similar three-dimensional structures and functions. However, structures show higher levels of conservation than sequences and the comparison of 3D structures is more useful in revealing functional relationships that are hidden at the primary sequence level.

Media
This picture was made using the MATRAS server 3D Matras Alignment
Learning from Structural Folds¶
The presence of a local structural motif can provide information about the functional role of a protein. In the case of the protein shown below, the presence of a "helix-turn-helix" motif in the folded structure of the protein hints at the ability of this protein to bind to DNA.
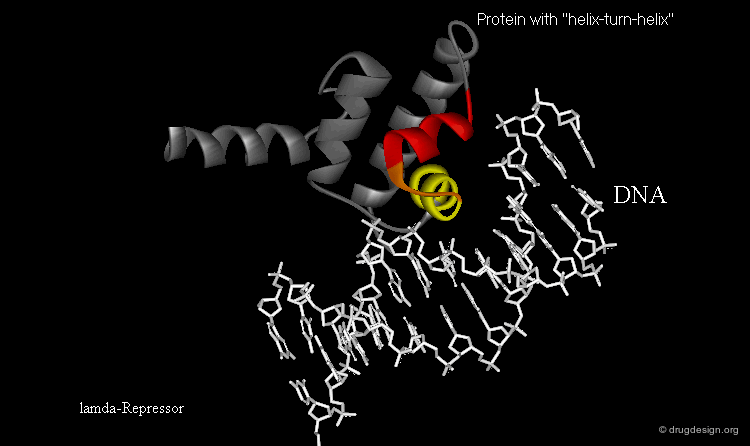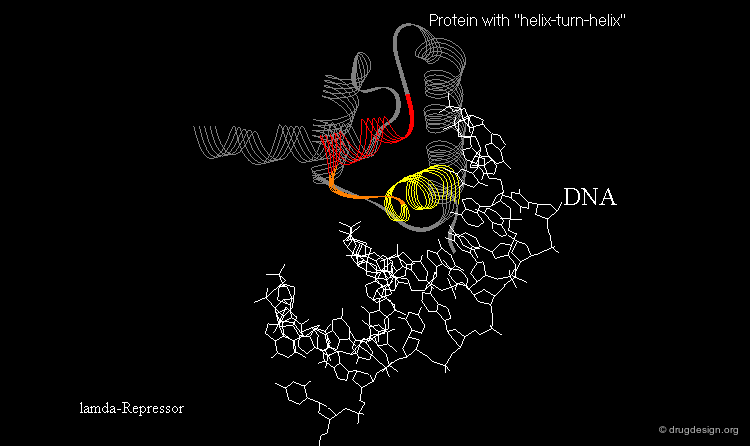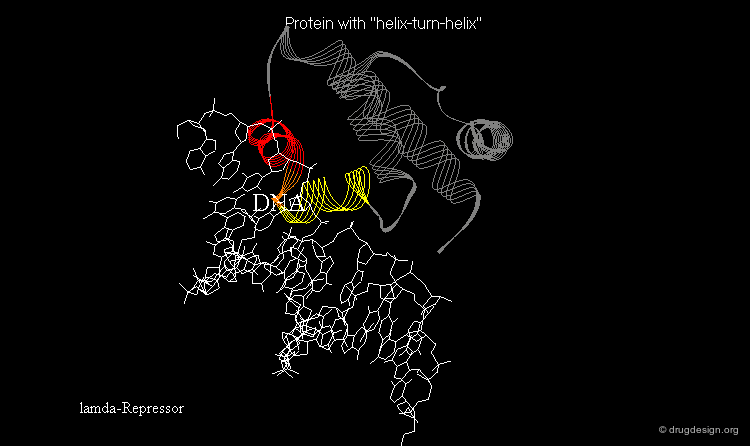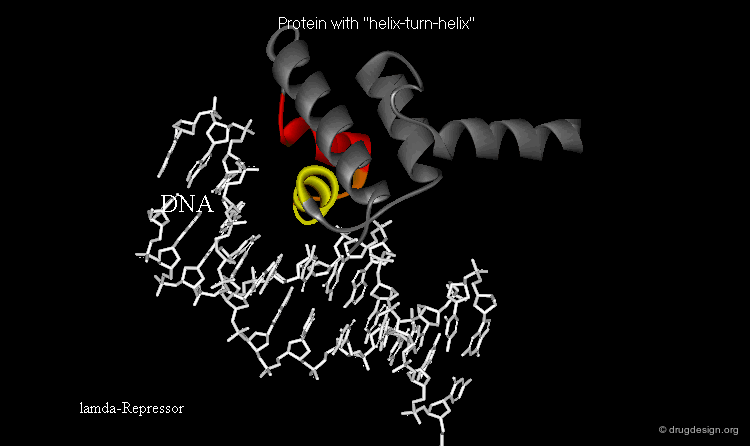
wikipedia
Learning from Molecular Shape¶
Biomolecules do not see and recognize each other as a set of atoms and bonds; rather they perceive shapes carrying complex forces. Analysis of molecular surfaces of proteins with their associated physico-chemical properties (such as electrostatic potentials etc...), can provide detailed information that is useful in understanding the properties and the function of these proteins. An important field of structural bioinformatics called 'molecular docking' aims at predicting computationally the association of two molecular surfaces.

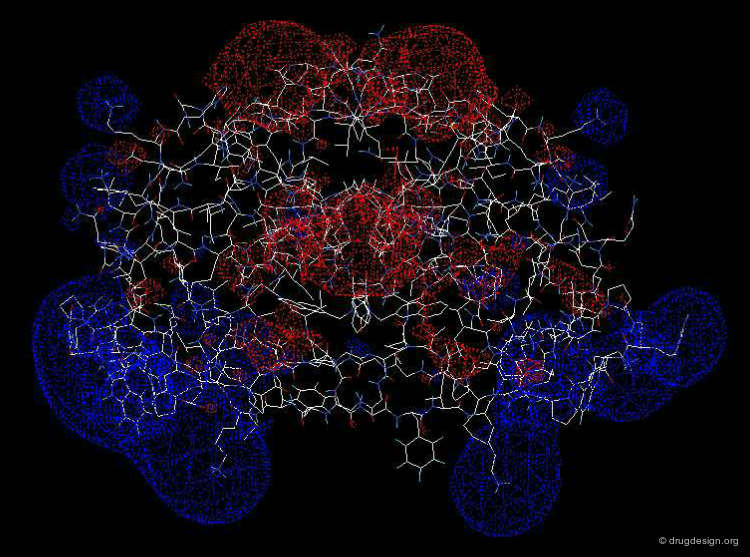
Media
This docking picture shows the binding of ubiquitin aldehyde with UBP Docking
The electrostatic potential was generated using the DeepView Swiss-pdb Viewer Electrostatic potential picture
Example of Knowledge Derived from 3D Structure¶
One of the first examples of 3D knowledge that led to a better understanding of life processes at the molecular level was the Watson and Crick DNA double helix model deduced from the X-ray photographs of DNA strands made by Rosalind Franklin in the early 1950s. This was a major step forward in charting many biological processes such as translation, regulation and replication that are carried out through the interaction of other molecules with DNA.

Is Structure Sufficient to Predict Function?¶
Proteins are not static; to fulfill their function they undergo a wide range of motion and conformational rearrangements. The knowledge of a static structure may not be sufficient to understand its biological function. An important component of structural bioinformatics is the development of experimental and computational methods (such as molecular dynamics) that sheds light on the dynamic behavior of biological systems. These methods simulate the way biomolecules behave, specifically recognize each other, interact and trigger biological events at the molecular level.
Exploiting Knowledge to Design New Drugs¶
The main application of structural bioinformatics is the discovery of novel therapeutic agents. By knowing the 3D architecture of a macromolecular system, rational drug design exploits the detailed features of recognition and discrimination that are associated with the specific arrangement of the chemical groups of a target macromolecule. New molecules are designed to interact optimally with the macromolecule to block or trigger a specific biological action.

wikipedia
Bridge between Genomics and Drug Discovery¶
Structural bioinformatics is the bridging discipline between bioinformatics (the genomics world) and cheminformatics (the drug discovery world). It is the key to the "gene-to-drug" dream and answers questions such as: "how is known genetic information converted into a therapeutically useful agent?" The principle of the approach is to translate both genomic and small molecule knowledge into 3D structural data that can then be exploited in the structure-based drug design world.
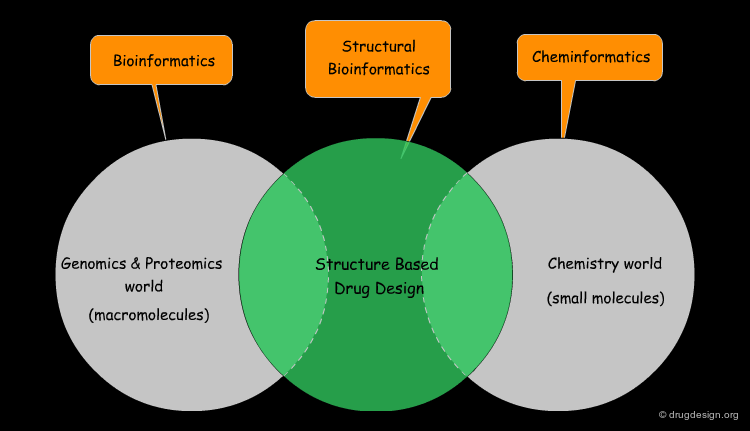
wikipedia
Tools Developed by Structural Bioinformatics¶
To achieve its goals structural bioinformatics focuses on the development of tools for: (1) encoding and visualizing biomolecules, (2) predicting the 3D structure of biomolecules, (3) storing, retrieving and searching in structural databases, (4) analyzing structural data, (5) performing data-mining, (6) calculating binding scores and (7) simulating molecular processes.

Architecture of Biomolecules¶
Biomolecules in the Cell¶
In this section we describe the architecture of the two most important biomolecules present in our cells: nucleic acids and proteins.

DNA/RNA Structure¶
Nucleic acids are essential materials found in all living organisms. Their main function is to maintain and transmit the genetic code; as such, they are the information carriers in the cells. This information is stored in the form of tremendously long polymer chains. Although the information they carry is one-dimensional, it is essential to understand the 3D structure of nucleic acids. This structure dictates their organization, functions and interactions with proteins.

wikipedia
DNA is the Genetic Material¶
DNA (Deoxyribo Nucleic Acid) is the genetic material of the vast majority of living cells. The language of the genetic code contains only 4 letters (A,T,G,C). Information about protein synthesis, which is the main machinery in the cells, is carried as shown in the table. Each triplet of nucleotides ("codon") encodes one amino acid. In the figure, click on one amino acid to visualize its corresponding codon.
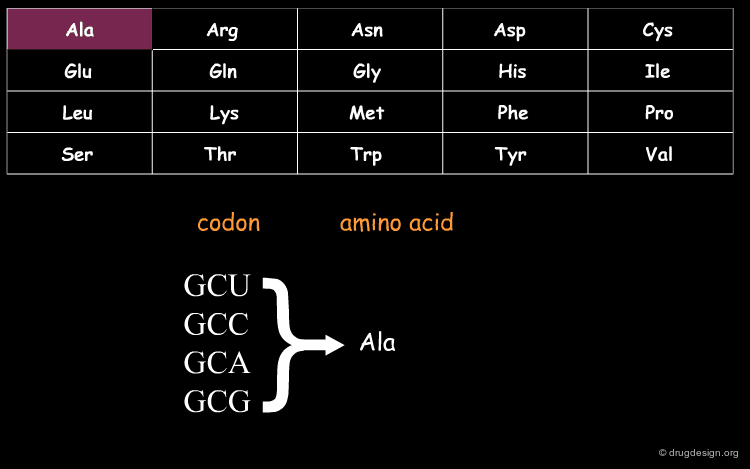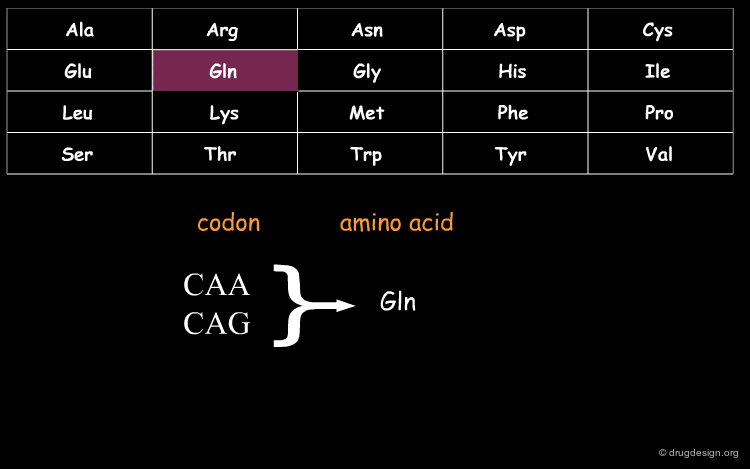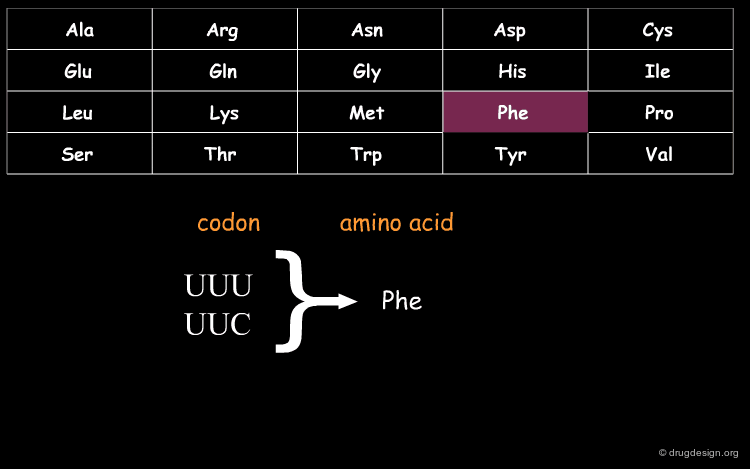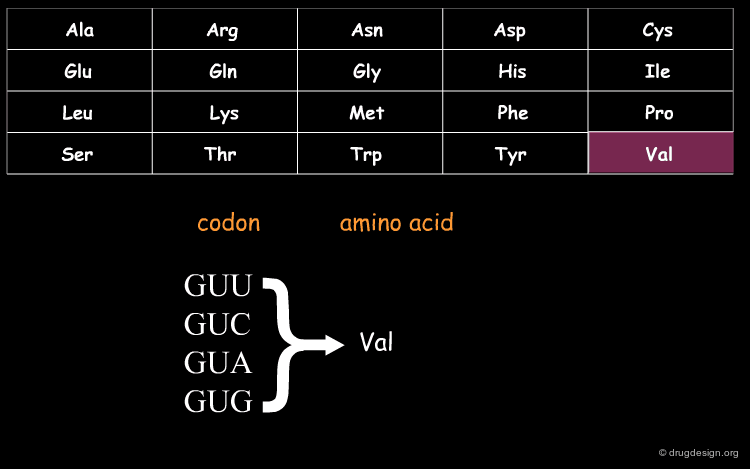
wikipedia
DNA Variability¶
The genetic code is extremely long (3x109 characters in humans), which is enough for the phenotypic variability seen in nature. Because of its length, the DNA molecules must be carefully packed and preserved. Many proteins in the cell are involved in these processes.

Media
This picture was made using the VMD and the Virtual DNA Viewer Extension VMD
Importance of the DNA 3D Structure¶
In order to understand how the DNA fulfills its function, it is necessary to understand its structure. In 1952, Hershey showed that DNA is indeed genetic material. In 1953 Watson and Crick, based on Rosalind Franklin's research, modeled the double helix structure of DNA. Their model accounted perfectly for the role of DNA as the genetic code and its ability to replicate. DNA folds into specific three-dimensional conformations, which are partially determined by the sequence of nucleotides.


The Building Blocks¶
Three types of chemicals make up the building blocks for nucleic acids: an aromatic base, a sugar ring and a phosphate group. The sugar and the phosphate constitute the non-specific backbone of the DNA. The base constitutes the specific part which actually holds the information and accounts for most of the interactions with other molecules.

Base¶
The group that gives each nucleic acid unit its specificity is the organic base. DNA contains two purine bases (adenine and guanine) and two pyrimidine bases (cytosine and thymine). Purine bases are made of two planar aromatic rings, whereas pyrimidine bases contain only one aromatic ring. In RNA the thymine base is replaced by uracil. Polar atoms in the ring or attached to the ring are capable of creating hydrogen bonds with polar atoms of other bases.

Sugar¶
The second chemical group in nucleic acids is the sugar. The sugars in DNA and RNA are pentoses (5 carbons). The ring of the sugar is flexible and non planar (in contrast to the rings of the base), and can adopt several conformations.

wikipedia
Phosphate¶
The inorganic acid H3PO4 (phosphoric acid) gives the nucleic acids an overall net negative charge. A basic DNA building block unit (nucleotide) contains three attached phosphate groups. However, in the polymerization of DNA two phosphates are released and one is incorporated into the DNA. Most of the interactions between the DNA and proteins which are not specific to the DNA sequence are with the phosphate groups.

wikipedia
Putting the Building Blocks Together¶
The three groups composing each unit of nucleic acids are connected by covalent bonds. The phosphate group is connected to carbon number 5 of the sugar. The sugar is connected to the base by a covalent single bond between carbon number 1 of the pentose ring and a nitrogen atom on the aromatic ring of the base. The assembly of these three groups defines a nucleotide. Nucleosides consist of the base and the sugar without the phosphate.

Nomenclature of Nucleotides and Nucleosides¶
Nucleotides are the monomers of nucleic acids. They may have one, two or three phosphate groups; the sugar can be ribose or deoxy-ribose and the base can be a purine or a pyrimidine base. Use the cursor to select a base, a sugar and a phosphate to visualize the corresponding nucleotide.

Nucleotides of Nucleic Acids¶
Mono-phosphate nucleotides are the structural units of nucleic acids. In DNA the sugar is always a deoxy-ribose. The nomenclature of the different nucleotides and nucleosides of DNA is shown in the table below.


The Double Helix Structure¶
In the early 1950s, Watson and Crick suggested that DNA was made of two helical strands of polynucleotides, one going up and the other going down. Their model proved to be correct and explained how, during cell division, the two strands separate and promote the assembly of two new strands identical to the ones existing before. This double helical structure enabled us to understand how the genetic information is maintained and how it is replicated during cell division.

wikipedia
DNA Helices are Antiparallel¶
Each strand of DNA has directionality. In other words, one edge is not identical to the other and there is no symmetry along the strand. The molecule has two different edges, termed the 3-prime edge and the 5-prime edge. The DNA molecule is arranged such that the two strands are lying next to each other but in opposite directions, such that the 3' edge of one strand is adjacent to the 5' of the other strand and vice versa. A phosphate group is always found in the 5' edge and hydroxyl group is found in the 3'. Polymerization occurs at the 3' edge.

Hydrogen Bonding Pattern¶
The two strands of the DNA are connected by hydrogen bonds. These forces are non-covalent interactions but are relatively strong and keep the two DNA strands in a tight association. Each nucleotide is capable of creating favorable hydrogen bond patterns with one other nucleotide. Adenine and Thymine interact with two hydrogen bonds. Cytosine and Guanine interact with three hydrogen bonds and therefore have a stronger association.


Aromatic Base Stacking¶
Another important interaction that stabilizes the DNA structure is termed stacking and refers to the interaction between adjacent parallel aromatic bases. This interaction results from sharing the electron clouds of the aromatic rings. The interaction is slightly stronger in purines, which possess a double ring.

wikipedia
Major and Minor Grooves¶
The two DNA strands are not straight; they form a helix. Due to its nature, this helix contains grooves which are preferred sites for proteins and other molecules to interact with the DNA. One significant groove in which the bases are highly exposed is termed the major groove. Most of the protein interactions occur at this site. The minor groove is located on the other side of the bases. It also serves for interactions with proteins, but less often.

DNA forms¶
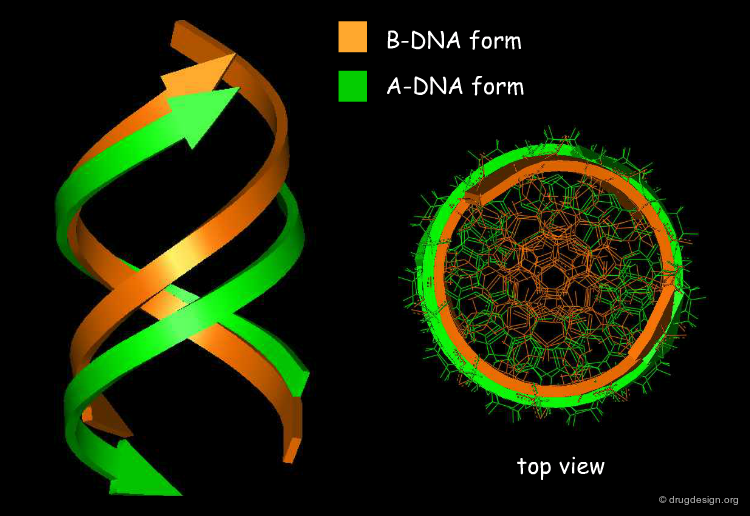
There are several forms of DNA double helices. The most common is the B-DNA. In every B-DNA cycle there are 10 base pairs; the distance between successive bases along the molecule axis is about 3.4 Å. In dehydrated environments, the DNA may appear as A-DNA. In every A-DNA cycle there are 11 base pairs so the molecule is more condensed, and accordingly the diameter of A-DNA is greater than that of B-DNA. Another uncommon form of DNA is the Z-DNA. The helix has an opposite rotation, composed of C and G nucleotides and is less symmetrical.

G-Quadruplex Conformation¶
In addition to the duplex DNA conformations, certain DNA sequences can fold into structures that are four-stranded. In particular Guanine-rich nucleic acid sequences are capable of adopting this type of organization, which is called G-quadruplex. The G-quadruplex structure is stabilized by hydrogen bonds between the edges of the bases and chelation with a metal (e.g. potassium) located in the center. Below the 3D and 2D structure of a G-quadruplex is illustrated.


book
Quadruplex Nucleic Acids Royal Society of Chemistry, Cambridge 2007
DNA versus RNA¶
There are several important differences between DNA and RNA molecules. (1) The sugar of RNA is always a ribose instead of a deoxyribose (with an extra hydroxyl group at carbon number 2 of the sugar), (2) the RNA molecule does not contain thymine bases; instead they are replaced by uracil, and (3) the DNA molecule normally appears as a double strand, while the RNA molecule appears as a single strand.

3D Folds of RNA¶
Due to its single strand structure, the RNA molecule is less stable than DNA. Base pairing might occur between bases along the single strand giving rise to various three dimensional folds. Note that in the structure below (hairpin fold) intra-molecular hydrogen bond interactions are formed between bases of the same single strand.


wikipedia
Protein Structure¶
Basic principles in protein structure are illustrated in this topic, which covers their amino acid building blocks and their folded, complex 3D architecture.

wikipedia
Media
This picture was made using KiNG (Kinetic Image, Next Generation) KiNG Home page
Proteins are Fundamental to Life¶
Proteins form the very basis of life. They are the most abundant macromolecules in living cells. Thousands of different proteins can be found in a single cell, and proteins have been identified in all parts of all cells. Proteins affect virtually every property that characterizes a living organism.
Structural Diversity of Proteins¶
The great diversity of the functional properties of proteins is directly related to their subtle three-dimensional structures. Every protein has a unique 3D structure that determines its function.

Importance of Protein 3D Structures¶
The formation of a protein in its biologically active form requires the folding of the protein into a precise three-dimensional structure. Changes in this structure (for example by denaturation) are consistently associated with loss of function of that protein.

Chemical Nature of Proteins¶
All proteins are linear polymers made up of various combinations of 20 amino acids, and are composed mainly of C, H, O and N atoms. The amino acid sequence that defines the protein is determined by the transcription of the genetic code. Remarkably, a cell can produce proteins with different properties and activities by assembling the same 20 amino acids in many different combinations and sequences.

wikipedia
Challenges in Understanding Protein Structure¶
Despite the exciting advances in our understanding of protein folding in the last decade, it remains an unresolved enigma. The prediction of the 3D structure of a protein from sequence data alone is a formidable challenge.

wikipedia
Media
This picture was released into the public domain Wikipedia
{kind=link}
Protein Structure Complexity¶
The most striking feature of the folded conformation of a protein is its complexity, revealing the organization and the relationships of thousands of atoms in three-dimensional space. We will ease our way into the confusing jumble of atomic coordinates, distances, torsions angles and interactions with a short overview of protein architecture.

The Four Levels of Protein Architecture¶
Protein structure can be decomposed into different levels that follow precise rules and contribute to its overall shape. This is called the protein architecture, which can be split in a hierarchy of four levels of structural complexity. These correspond to the primary, secondary, tertiary and quaternary levels of protein structure.

Primary Structure¶
The first and most fundamental level of a protein is its primary structure, which describes the atomic connectivities of the protein. The primary structure is defined by the amino acid sequence and may include cysteine bridges that are formed during cross-linking.

wikipedia
Secondary Structure¶
The next level of protein structure, the secondary structure, includes local regular conformations of the polypeptide chain. These are dictated by the hydrogen bonds of the biopolymer creating specific secondary structures that are energetically favored in the conformational Φ-Ψ space of the protein. The two major elements of the secondary structure are the alpha-helix and the β-sheet.


wikipedia
Tertiary Structure¶
The tertiary structure of a protein is the overall 3D architecture of the folded polypeptide chain, which also includes the assembly of the various secondary structure elements in 3D.

wikipedia
Quaternary Structure¶
Finally, proteins consisting of more than one polypeptide chain display a quaternary structure, which refers to the spatial relationship between the individual polypeptide chains (subunits).

wikipedia
Biomolecular Properties¶
Protein Flexibility and Motion¶
Proteins were considered to be static objects for a long time. This static view did not take into account the true dynamic nature of bio-molecules that are in constant movement, making motions of diverse amplitudes and time scales, from the level of individual atoms up to the level of structural domains. These motions are crucial to the biological functions of proteins.

articles
Einfluss der Configuration auf die Wirkung der Enzyme Fischer E. Ber. Dt. Chem. Ges. 27 1894
wikipedia
Importance of Dynamic Motions in Biological Processes¶
Molecular motions can have two distinct roles. First they allow biomolecules to interact and adapt to other biomolecules (induced-fit), and second they dictate the biochemical or mechanical function of macromolecules. This function is generally only one small part of a larger molecular cell program. Examples of induced-fit and functional roles are given in the next pages.
wikipedia
Example of Function: ATP Synthase¶
ATP Synthase is the universal carrier of energy in the cell. The enzyme catalyses the synthesis of ATP from ADP. ATP biosynthesis results in the production of a complex molecular machine composed of two connected rotary motors, where each motor is powered by a different fuel. Animation of ATP Synthase can be seen by activating the link appearing in the "additional information" panel (on the left side) which illustrates the importance of molecular motions in biological processes.

wikipedia
Media
From Wikipedia, the free encyclopedia, This picture has been released into the public domain by the copyright holder ATPsynthase Picture
{kind=link}
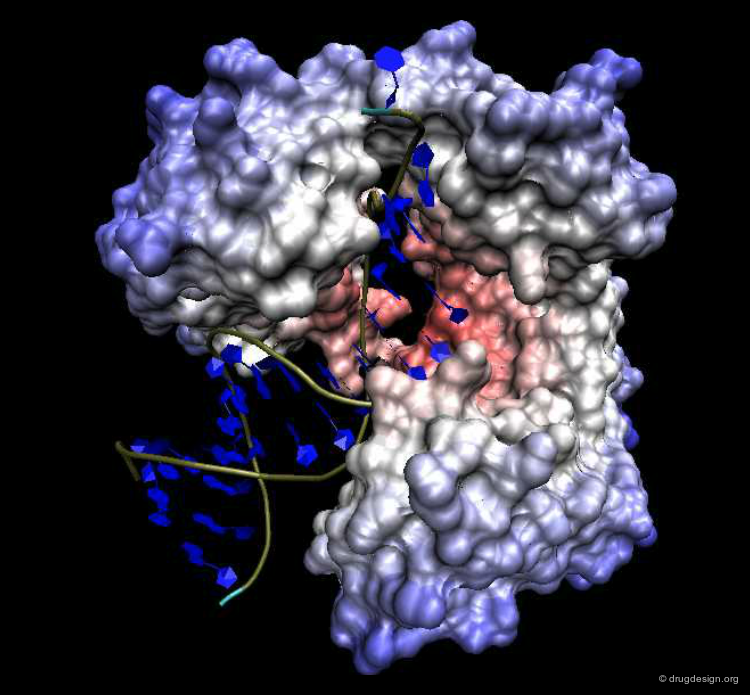
Example of Function: DNA Biosynthesis¶
Another example illustrating the importance of protein motion is DNA polymerase, an enzyme that catalyses DNA biosynthesis. This is a highly complex dynamic process where DNA polymerase assists DNA replication. In the figure below the structure of the DNA polymerase is shown both at a 3D and a schematic level.
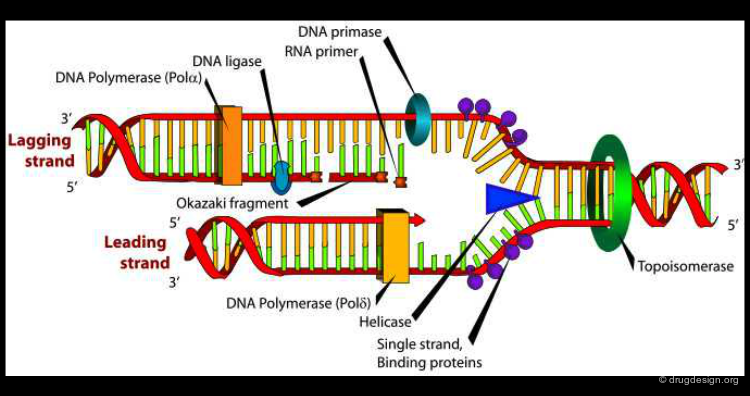
wikipedia
Media
The schematic representation image has been released into the public domain by its author DNA replication picture
{kind=link}
Example of Function: Molecular Switch¶
The differences in the arginine kinase shown below in two different conformational states result from the motion between four rigid groups of non-sequential residues, a change that acts as a molecular switch that controls cellular processes: switch "on" (blue) and switch "off" (red). The origin of the conformational changes induced in the different parts of the enzyme are the outcomes of the interactions with both ATP and the substrate of the kinase.
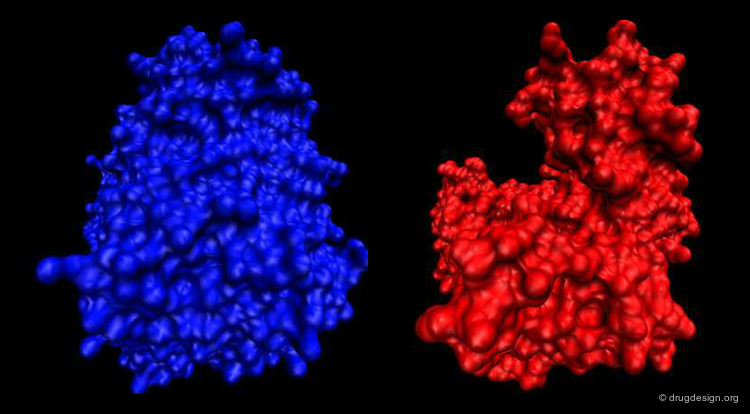
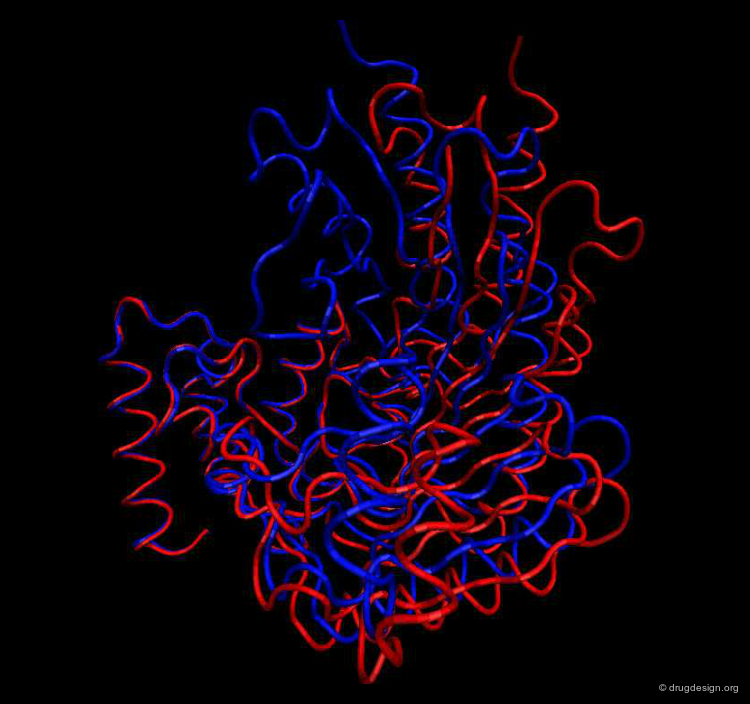
articles
Induced fit in guanidino kinases-comparison of substrate-free and transition state analog structures of arginine kinase Yousef M.S. et al. Protein Science 12 2003 10.1110/ps.0226303
Example of Induced-Fit: RNA-Protein Recognition¶
Studies of the interactions between RNA and proteins reveal that the formation of almost every RNA-Protein complex involves conformational changes in the protein, the RNA, or both ("mutually induced-fit"). In the example below the complex formed between RNA and the U1A protein is shown as well as the structures of the free molecules. In this example both molecules undergo important conformational changes.

articles
Induced fit in RNA-protein recognition Williamson J.R. Nature Structural Biology 7 2000 10.1038/79575
Example of Induced-Fit: Ubiquitous Proteins¶
Many proteins are ubiquitous and can bind to different protein targets. This is the case for Calmodulin, which undergoes conformational changes upon binding to calcium and can then bind to a number of specific proteins to elicit a specific response.
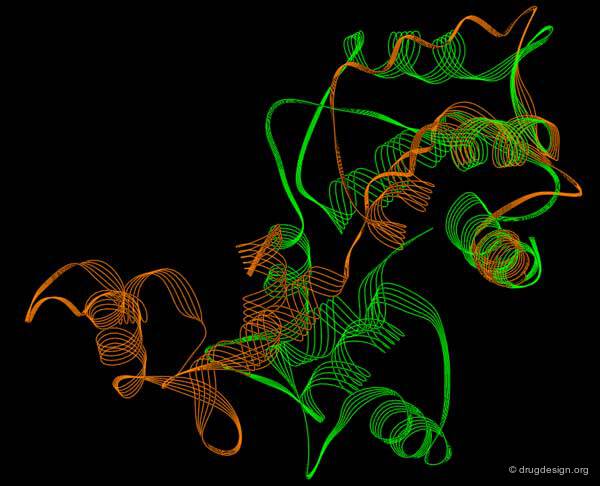


Types of Molecular Motions¶
Protein flexibility has different levels. (1) Individual atoms exercise very high frequency vibrations but are usually not very important for the function of the protein. (2) The rotation around single bonds are important in terms of the function of the protein because they lead to collective motions of several residues such as loop changes or side chain rotations; they accompany conformational changes such as those occurring in the vicinity of binding sites. (3) Important conformational changes associated with the relative motions of large structural units; they are extremely important for protein function.
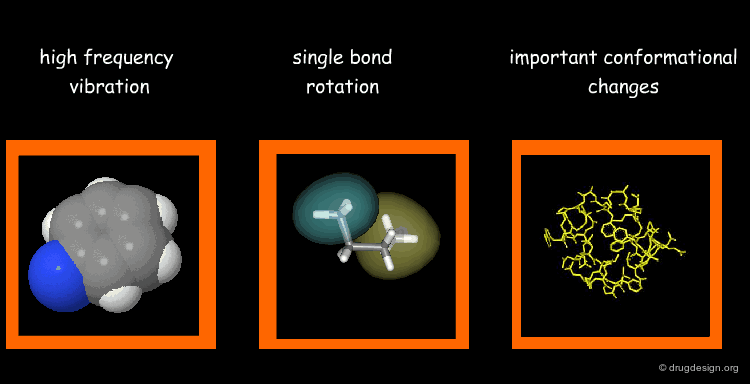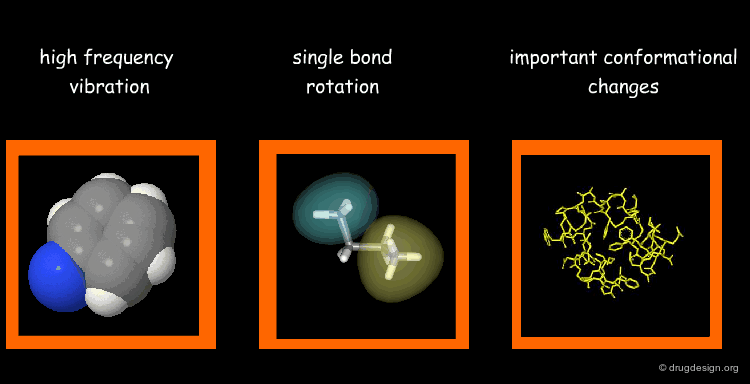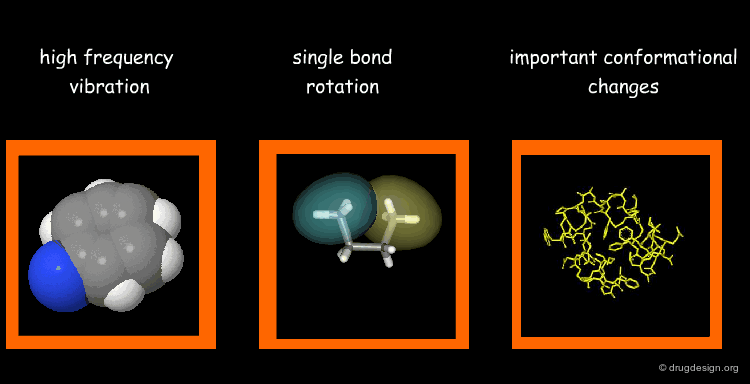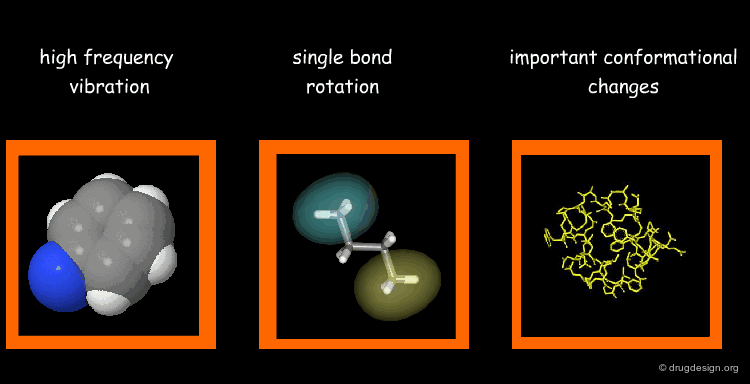
Time Scale of Protein Motion¶
Illustrated below are the time scales for the different types of motions previously mentioned. Protein motions occur at time scales in the range of pico to milli-seconds. A motion starts with the side chains that change their rotameric state. It continues to loop movements, hinge bending and subunit displacements, respectively.

Methods to Study Protein Motions¶
There are many methods to study protein motions ranging from experimental techniques to simulation methods with computers. A brief outline of these methods is given in the following pages.

Experimental Techniques to Study Protein Motions¶
Experimental 3D structure determination methods provide some indications as to macromolecular dynamics. X-ray crystallography assigns a value for the fluctuation of each atom. This parameter is known as the B-factor (temperature-factor) that expresses the mobility/uncertainty in the position of the corresponding atom. NMR spectroscopy, with measures of relaxation times and order-parameters tells us about the dynamics of the molecules. Other methods such as Mossbauer spectroscopy, neutron scattering and hydrogen exchange can also be used for studying protein mobility. Another way of acquiring information about the dynamics of a protein is to superimpose the different X-ray structures solved for a protein (i.e. complexes made with several inhibitors).

Simulation Methods to Study Protein Motions¶
Contrary to experimental techniques that only provide an average state of the sample, molecular simulations can deliver useful insights and precise details on the dynamics of each component of the protein ranging from the movement of single atoms to fragments, loops and domains. Molecular dynamics is the most commonly used simulation technique for studying a protein alone or interacting with other molecules. MD is a statistical mechanics method that follows the time course by integrating equations of motion. This method is described in some detail in the section "Molecular Dynamics".

Normal Mode Analyses (NMA)¶
"Normal mode analyses" is a method widely used in IR and Raman spectroscopy to analyze vibrational modes that can be used to study protein conformational changes. The method attracted renewed interest after the recent discovery that about 50% of protein movements can be modeled using a small number of low-frequency normal modes. Typically, it is used for the study of collective motions of complex biological systems.

Media
This picture was made using the WEBnm@ server (pdb:1hvr) Normal Mode Analysis
Molecular Dynamics vs Normal Mode Analyses¶
In principle, Molecular Dynamics (MD) is better than Normal Mode Analyses (NMA) for the simulation of protein motions: the harmonic approximation introduced in NMA restricts the search to a small region of the conformational space. However, it has been observed that NMA produces reasonable results for a great number of systems. Moreover it has the advantage of being much less costly in terms of computing time than MD. For these two reasons NMA has become the preferred method for routine studies of protein motions aimed at describing protein dynamics in a concise way.

Database of Macromolecular Movements¶
Marc Gerstein's group has compiled a database of macromolecular movements with associated tools for flexibility and geometric analysis. Conformational changes of more than 240 individual protein motions are described and visualized by movies. A web-based tool called the "Morph Server" enables the user to generate, interpolate and animate the transition between two distinct states of a macromolecular system (described as PDB files).

wikipedia
Assembly of Biomolecules¶
Biological Molecule Association¶
Every process in living cells evolves as the result of a complex association between molecules. Self assembly mainly occurs by specific molecular interactions. The ability to understand the nature and the driving forces behind these interactions is crucial in the life sciences.

articles
A confederacy of bunches: Fundamentals and applications of a self-associating protein Wandless T. J. Proc Natl Acad Sci USA 20(13) 2000
Molecular Recognition¶
Biochemistry reveals to us how inanimate molecules that constitute living organisms recognize each other to maintain and perpetuate life. Understanding how molecules recognize each other is referred to as molecular recognition and is presented in the following pages.
articles
Molecular Docking: A Problem With Thousands Of Degrees Of Freedom M. Teodoro and G. Phillips and L. Kavraki IEEE Press
2001
wikipedia
The Recognition Process¶
Molecular recognition is a multistep process. It begins by the approach of the molecules followed by desolvation of the interacting surfaces. The ligand starts to penetrate the active site of the protein. Orientational and conformational changes take place to form favorable interactions such as new hydrogen bonds, hydrophobic and electrostatic interactions. As a result, specific perturbations of the receptor induce the biological response.

Complementary Features Upon Binding¶
One of the essential principles in molecular recognition is the structural complementarity between interacting molecules (known as the "lock-and-key" model). This was postulated in 1894 by Emil Fisher when he observed that invertase from beer yeast hydrolyzed α-glucosides but not β-glucosides, while emulsin hydrolyzed the β- but not the α-glucosides.

Role of Native Protein Configuration¶
In 1936 Mirsky and Pauling demonstrated that the specific properties of native proteins were due to their uniquely defined configurations, whereas denaturated proteins lose these properties by lacking such organization. This idea was introduced twenty years before Anfisen's experiments on ribonuclease refolding and the X-ray determinations of the structure of myoglobin that finally led to the acceptance that highly stereospecific 3D structure is a fundamental requirement for protein function.

articles
On the structure of native, denatured and coagulated proteins Mirsky AE, Pauling L Proc. Natl. Acad. Sci. USA 22 1936
wikipedia
Tolerance Upon Binding¶
In 1950 Karush observed that many ligands of different shapes exhibited competitive binding to serum albumin. He extrapolated the lock and key principle and proposed that there is some tolerance upon binding, a process that favors the best-fitting member from an ensemble of structures in equilibrium. He called it "configurational adaptability".

articles
Heterogeneity of the binding sites of bovine serum albumin Karush F J. Am. Chem. Soc. 72 1950
Intrinsically disordered protein Dunker A. K. et al. Journal of Molecular Graphics and Modelling 19 2001 10.1016/S1093-3263(00)00138-8
Media
This picture was made using KiNG (Kinetic Image, Next Generation) KiNG Home page
The "Induced-Fit" Theory¶
Independently, Daniel Koshland introduced the "induced-fit" theory in 1958. This theory stipulates that in the recognition process, protein and ligand mutually adapt themselves by small conformational changes until an optimal fit is achieved. This model accounts for molecules that can bind despite having non-perfect complementarity in the unbound state.


articles
Remembering Daniel E. Koshland Jr. (1920-2007) Dahlquist F.W. Protein Science 16 2007
Complexes of alkylene-linked tacrine dimers with Torpedo californica acetylcholinesterase: Binding of bis(5)-tacrine produces a dramatic rearrangement in the active-site gorge Rydberg, E.H., Brumshtein, B., Greenblatt, H.M., Wong, D.M., Shaya, D., Williams, L.D., Carlier, P.R., Pang, Y.-P., Silman, I. and Sussman, J.L. J. Med. Chem. 49 2006
eMovie: A storyboard-based tool for making molecular movies Hodis, E., Schreiber, G., Rother, K. and Sussman, J.L. Trends in Biochemical Sciences 32 2007
wikipedia
Example of Enzyme Adaptation to Inhibitor Binding¶
An example of enzyme adaptation to inhibitor binding is described in the work of Gee et al (2007) on phenylethanolamine N-methyltransferase (PNMT), the enzyme that catalyzes the N-methylation of norepinephrine. They found that some inhibitors (such as SKF 29661) can substantially increase the size of the active site of this enzyme.
![]()
articles
Enzyme Adaptation to Inhibitor Binding: A Cryptic Binding Site in Phenylethanolamine N-Methyltransferase Gee C,L. et al. J. Med. Chem. 50 2007 10.1021/jm0703385
wikipedia
Example of Ligand Adaptation upon Binding¶
It is important to note that induced-fit adaptations not only apply to conformational changes of the receptor but also to those of the ligand. This is illustrated below in the binding of acetylcholine to the nicotinic acetylcholine receptor. The 3D structure of acetylcholine in its free solution-state conformation (left) is compared to the conformation of acetylcholine in its receptor-bound state (right).
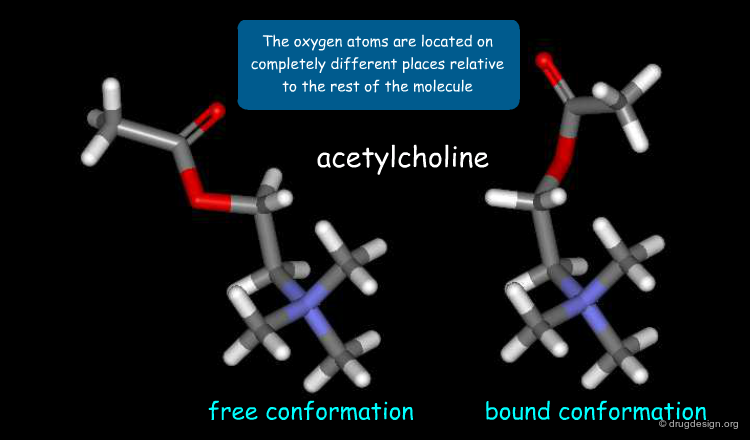
articles
Conformation of acetylcholine bound to the nicotinic acetylcholine receptor RONALD W. BEHLING, TETSUO YAMANE, GIL NAVON, AND LYNN W. JELINSKI Proc. Nati. Acad. Sci. USA 85 1988
wikipedia
Maximizing Surface Contacts¶
The aim of the induced-fit process is to maximize the common shared area of the interacting molecules. This phenomenon, usually referred to as the plasticity of the protein surface, contributes to the thermodynamic stability of the complexes. To better illustrate the 3D complementarity between the two proteins shown below, a "slab" option was used that shows cross sections of the system.

Motions Associated to Induced-Fit¶
Motions associated to the induced-fit process include small backbone displacements and side-chain and loop rearrangements. Adaptation of the binding site to the shape of the ligand is mainly achieved through rearrangement of the side chains of the protein that can change their rotameric state with small energy changes. It has been observed that 40% of all side chains of a protein have a 10%-40% chance of undergoing a substantial rearrangement upon binding of the ligand.
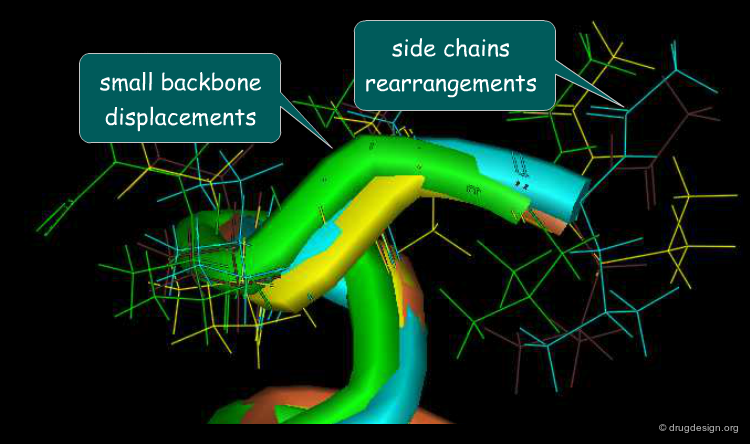
articles
Protein Flexibility and Drug Design: How to Hit a Moving Target Carlson, H.A. Current Opinion in Chemical Biology 6 2002
Experimental Evidence of the Induced-Fit Model¶
In a work designed to provide experimental evidence for the induced-fit model Gutteridge and Thornton analyzed the X-ray structures of 60 different enzymes to see whether conformational changes could be observed between the apo and the complexed forms. They found that induced-fit motions in most enzymes were very small (less than 1 Å RMSD across the whole protein). This confirms the validity of the induced-fit theory and demonstrates that small conformational changes have a major impact on the molecular recognition process.

articles
Conformational changes observed in enzyme crystal structures upon substrate binding GUTTERIDGE A, THORNTON J: J. Mol. Biol. 346(1) 2005 10.1016/j.jmb.2004.11.013
Incorporating protein flexibility into docking and structure-based drug design Barril X, Fradera X Expert Opin. Drug Discov. 1(4) 2006 10.1517/17460441.1.4.335
Large Rearrangements¶
In addition to small induced-fit motions, larger rearrangements can be observed involving the relative motions of domains or substructural parts. This phenomenon was first observed in 1978 by Bennett and Steitz in the binding of glucose with hexokinase. The free enzyme is shown here in blue and the complex with glucose appears in red. Note the rearrangement that enabled the substrate to bind.

articles
Glucose-induced conformational change in yeast hexokinase Bennett, W.S., Jr. and Steitz, T.A. Proc. Natl. Acad. Sci. USA 75 1978
Structural dynamics of yeast hexokinase during catalysis W.S.Bennett and T.A.Steitz Philos Trans R Soc Lond B Biol Sci 293(1063) 1981
A refined model of the sugar binding site of yeast hexokinase B. Anderson CM, Stenkamp RE, McDonald RC, Steitz TA. J Mol Biol 123(2) 1978
Media
This picture was made using the HingeProt server and VMD HingeProt Server
Role of Large Rearrangements¶
Large rearrangements are often key determinants in the mechanism and the function of a protein. For example the interconversion between the closed-inactive and the open-active conformations of an enzyme may enable the binding of the substrate or the release of a product.

Media
This picture was made using the HingeProt server and VMD HingeProt Server
The Domino Effect¶
In a multi-subunit protein a conformational change in one subunit can affect the conformation of the other subunits. The binding of a ligand to a protein may be regulated through specific interactions with one or more additional ligands. For example, these other ligands may cause conformational changes in the protein that affect the initial binding of the first ligand; this is the so-called "domino effect" sometimes observed in protein binding.

Proteins Described as Ensemble of Conformations¶
To mimic the plasticity inherent to all proteins which enables them to interact with and adapt to other proteins, a protein can be described as an ensemble of very different conformations. Moreover, a ligand can recognize and bind to any conformation and subsequently, the induced-fit mechanism occurs to maximize the corresponding binding.

articles
FOLDING FUNNELS AND BINDING MECHANISMS BUYONG MA ; KUMAR S. ; TSAI C.-J. ; NUSSINOV R. Protein eng. 12(9) 1999
Energy Landscape of a Protein¶
The profile of the potential energy surface of a protein is therefore represented by a great number of funnels, each one corresponding to a family of conformations, of which only a small number play a biological role and the remainder merely reflect the plasticity of the protein.
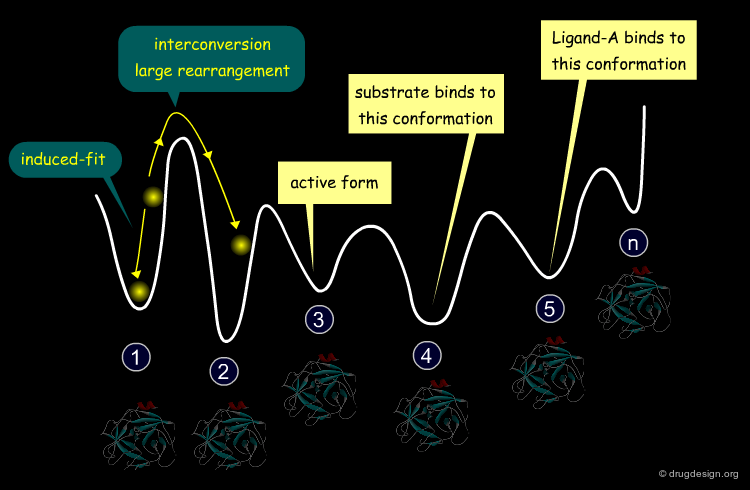
articles
FOLDING FUNNELS AND BINDING MECHANISMS BUYONG MA ; KUMAR S. ; TSAI C.-J. ; NUSSINOV R. Protein eng. 12(9) 1999
IMPLICATIONS OF PROTEIN FLEXIBILITY FOR DRUG DISCOVERY Simon J. Teague Nat Rev Drug Discov 2 2003
Conformational Selection Operated by a Ligand¶
The recognition process, in which the ligand binds selectively to a given conformation of the protein, is called "conformational selection". The conformational selection operated by the ligand increases the population of the selected conformation of the protein with respect to the other conformations and leads to the so-called "energetic induction" phenomenon.

articles
IMPLICATIONS OF PROTEIN FLEXIBILITY FOR DRUG DISCOVERY Simon J. Teague Nat Rev Drug Discov 2 2003
Energetic Induction Upon Binding¶
The binding of a ligand to the protein results in an energetic induction phenomenon where the potential surface of the protein and the preference between the conformational minima are altered.

Forces Involved in Molecular Recognition¶
Molecular interactions are responsible for the assembly of biological structures. A correct understanding of the forces that govern such interactions is important for gaining insight into molecular associations. These forces include Van der Waals interactions, electrostatic interactions and the formation of hydrogen bonds.

wikipedia
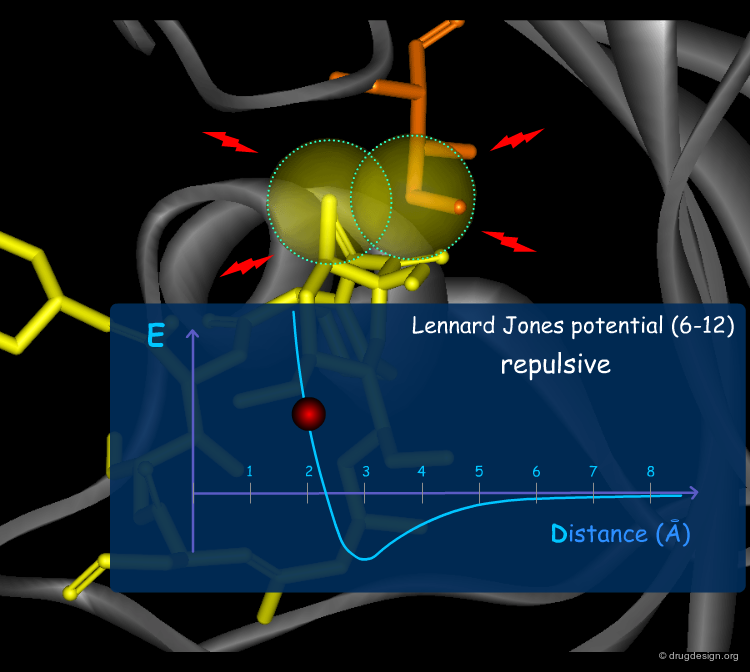
Van der Waals Forces¶
Van der Waals (VdW) forces between atoms are due to temporary atomic induced polarization between interacting atoms ("induced dipoles"). At long distances, individually, these forces are very weak, but the accumulation of a large number of such weak forces substantially contributes to a molecular association. At short distances, the van der Waals forces become strongly repulsive because of the steric clash occurring with the overlap of the electron clouds. The Lennard Jones (6-12) potential describes these two types of VdW interactions.


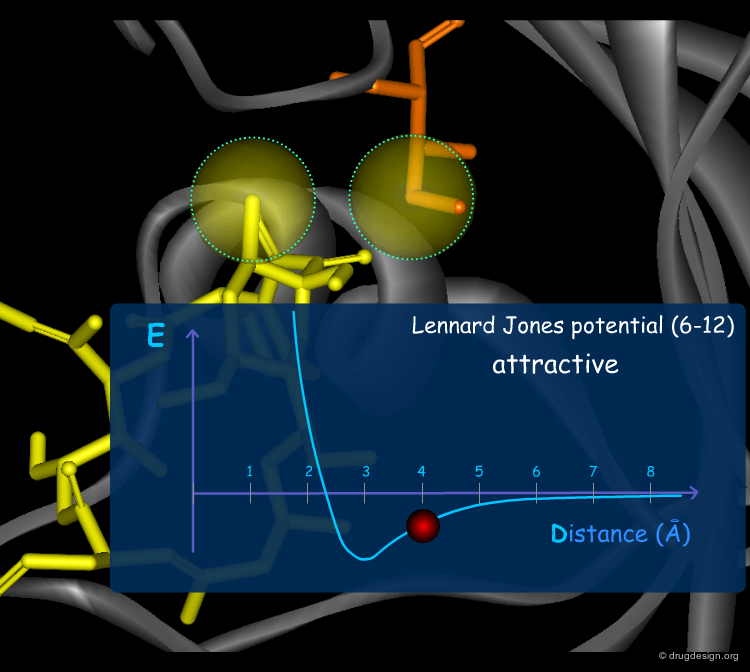


Electrostatic Interactions¶
Electrostatic interactions are strong forces acting between charged particles. They can be attractive (if the two charges are unlike) or repulsive (if the charges are like). The electrostatic potential can be simply described by Coulomb's law. It decays slowly as a function of the distance between the charges and therefore can still be meaningful at relatively long distances. In order to accurately describe the electrostatic potential it is preferable to locate partial charges and not only the net charge of the atoms.

wikipedia
Hydrogen Bonds¶
The hydrogen bond is perhaps the most significant non-bonded interaction in biology. It accounts for the double helix organization of two complementary DNA strands and also plays an important role in all types of macromolecules. For example, C=O---NH hydrogen bonds of the backbone of a protein account for the organization of its secondary and tertiary structure. Typically the interaction occurs between one hydrogen bond donor (e.g. NH, OH) and one acceptor (e,g. O, N) that share the hydrogen between them in a well- defined geometry.

wikipedia
Solvent Effect¶
The majority of molecular interactions occur in the liquid phase; therefore one must consider the solvent (often water) and its interactions with the surrounding molecules. In particular energies need to be expended for moving the polar part of a ligand out of the water and into the binding site.

The Role of the Solvent¶
Solvent molecules play many roles. First, there are direct interactions between solvent and solute molecules. Moreover, apolar molecules tend to aggregate in the presence of water. This effect is known to be entropically rather than enthalpically driven and is known to play a major role in dictating the folding pattern of proteins. Solvent molecules can also screen electrostatic interactions in the medium or form intermolecular bridges between a ligand and a protein.
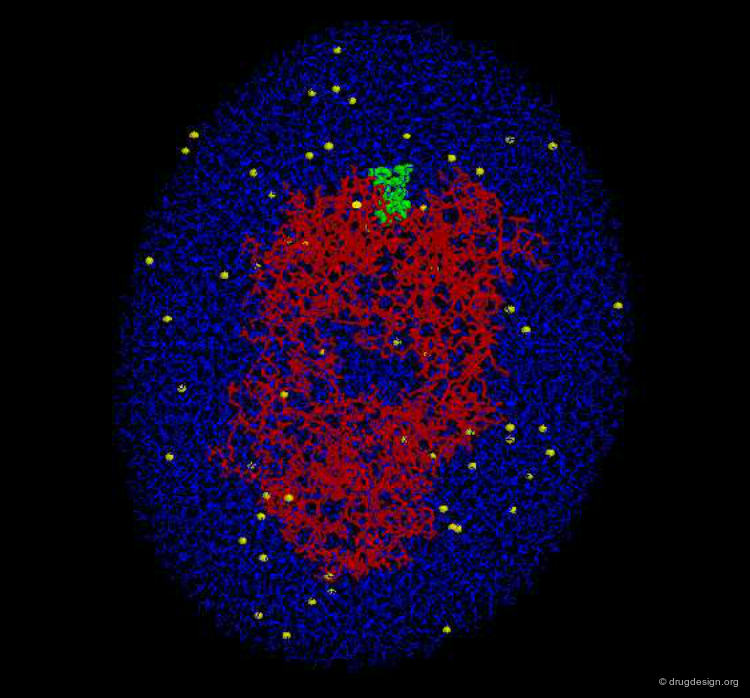
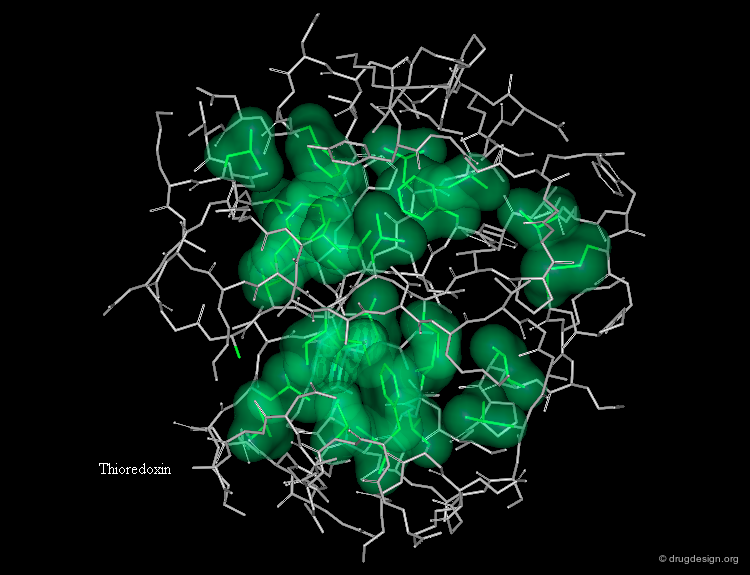
The Hydrophobic Effect¶
The aqueous solvent plays a crucial role in determining the conformational preferences and interaction forces of ligands and receptors. In particular, the hydrophobic effect (also called hydrophobic collapse) is caused by the unfavorable interaction of non-polar groups with water molecules; they tend to cluster together to exclude water and minimize the area of interaction with water. The interpretation of this phenomenon is that water-water interactions are the most favorable interactions and in effect, the water squeezes out the hydrophobes so it can interact with itself, leaving the hydrophobes to interact with themselves.
The Entropic Effects¶
The free energy of a system is composed of two components: enthalpy (or potential energy) and entropy (or "degree of disorder"). The solvent controls the entropy of a biological system by influencing the conformations of the interacting molecules. A flexible molecule has a better chance of finding an optimal fit into a receptor, but this is achieved at the cost of large conformational entropy.

articles
Ligand configurational entropy and protein binding Chang C.A et al. PNAS 104 2007
Enthalpy-Entropy Compensation¶
Many aspects of complex formation are unfavorable as they result in the loss of conformational degrees of freedom for the interacting molecules; therefore, highly favorable enthalpic contacts must compensate for this entropic loss.

wikipedia
Assessing Binding Interactions¶
Now that the forces that dictate molecular associations are well characterized, we can quantitatively assess them. This will pave the way to molecular simulations and the prediction of binding interactions and binding energies.

Free Energy of Binding¶
Binding energy is the energy required to disassemble a whole system into separate parts; it represents the mechanical work which must be done by acting against the forces which hold the molecules together. A complex has a lower energy than its constituent parts. Estimating the binding energy is a central issue in biology and structural bioinformatics.

wikipedia
Importance of Free Energy of Binding¶
From a chemist's point of view, free energies (also called Gibbs energies ΔG) are of utmost importance because differences in free energies determine binding constants, equilibrium rates, conformations and many other properties of a system. ΔG can be obtained experimentally or theoretically. Experimentally, this requires the availability of the two interacting molecules; theoretically, this is not needed at all, however they often need to approximate ΔG by the potential energy of the system.

Experimental Measures of Binding Affinities¶
The association of two biomolecules is an equilibrium process. The binding to a receptor is a phenomenon where the two molecules associate and dissociate, a process regulated by the law of mass action. A measure of the "goodness of fit" of a complex is measured by the Kd constant (dissociation constant) of the equilibrium. The dissociation constant has molar units (M); a nanomolar complex (Kd ~ nM) is more tightly bound than a micromolar one (Kd ~ µM).

wikipedia
Titration Curve to Measure Kd¶
The dissociation constant corresponds to the concentration of a ligand required to occupy 50% of its binding site on a particular protein. Experimentally, the dissociation constant of a system is measured by plotting the titration curve giving the percentage of occupation of the protein Θ as a function of the concentration of the ligand [L]. Kd is obtained by finding the point for which Θ = 50% in the graph.

wikipedia
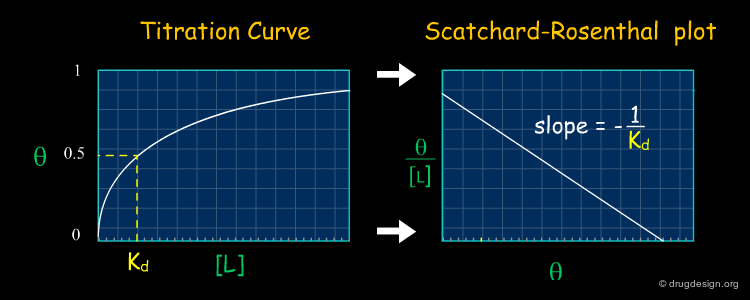
Scatchard-Rosenthal Plots¶
A convenient way to obtain Kd consists of linearizing the titration curve obtained on the previous page and plotting Θ/[L] as a function of Θ. This is the so-called Scatchard-Rosenthal plot in which Kd is the negative reciprocal of the slope. With such plots, it is easy to compare two sets of data; if the slopes for both sets are similar, the Kd will be similar.
wikipedia
Conversion of Kd into Energies¶
The relationship between the free energy and Kd is given by the equation ΔG = -RT LogKd. In this equation, R is the ideal gas constant, T is the temperature in Kelvin and Kd is the dissociation constant. Kd constants can be converted into energies, and vice versa. The following diagram represents ΔG (in kJ/mol) as a function of log Kd at room temperature.
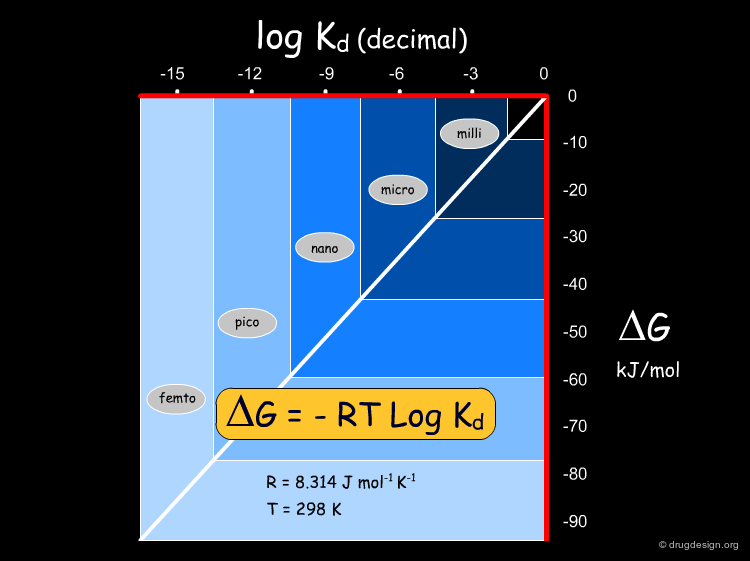
Theoretical Prediction of Binding Energies¶
The theoretical prediction of the binding energy of a system is of central importance for understanding biological mechanisms and this is also exploited in drug discovery. For example it enables a better understanding of why one ligand is preferred to another, and provides efficient means to speed the discovery of new molecules that bind tightly and specifically to particular proteins.
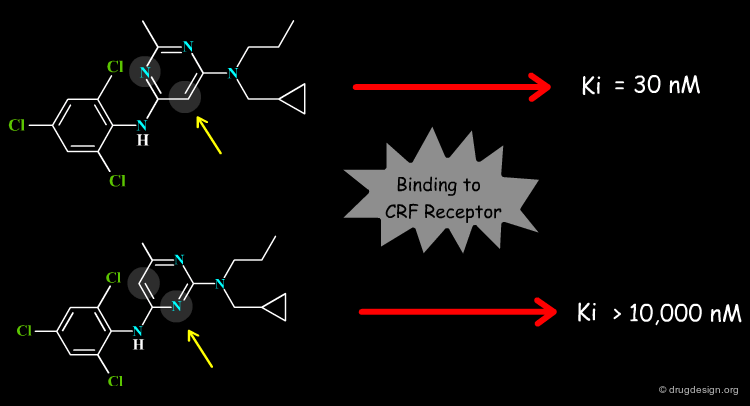
articles
Design and Synthesis of a Series of Non-Peptide High-Affinity Human Corticotropin-Releasing Factor1 Receptor Antagonists Chen Chen, Raymond Dagnino, Jr., Errol B. De Souza, Dimitri E. Grigoriadis, Charles Q. Huang, Kyung-Il Kim, Zhengyu Liu, Terry Moran, Thomas R. Webb, Jeffrey P. Whitten, Yun Feng Xie, and James R. McCarthy J. Med. Chem. 39 1996 10.1021/jm960149e
Solving the Schrodinger Equation¶
From a theoretical point of view quantum mechanics and its associated Schrodinger equation gives the exact description of a system and enables the calculation of energies, energy levels, wave functions and many other properties. It can be solved exactly for systems of 2 or 3 atoms, whereas for systems of fewer than 100 atoms, although it can be still used, approximations need to be introduced. The calculation of macromolecular binding energies with quantum mechanics is far beyond the current state of the art in this field.

wikipedia
Media
The Schrodinger picture is from the Wikimedia Commons (Public Domain) Erwin Schrodinger
{kind=link}
Adapted from K. Wilson UCSD The Schrodinger Equation
{kind=link}
Molecular Mechanics¶
Molecular mechanics remains an alternative for calculating the binding energies of large molecular systems. The method considers atoms as spheres, centered at the position of their nucleus and applies the laws of classical mechanics to calculate the potential energy of the system. Molecular mechanics gives no information about absolute free energies; however, it is useful for comparing the relative energy of two different configurations.

wikipedia
Force-Field¶
A "force-field" consists of a set of mathematical equations that simulate intra and inter-molecular forces which enable the calculation of the potential energy of a system. Forces and energies are directly related: the force is the negative derivative of the potential energy. Each component of the force-field models a particular force applied to the system. The sum of all these forces include stretching, bending, torsional, van der Waals, hydrogen bonds and electrostatic energies, and their total is an approximation of the total free energy ΔG that governs the system.

wikipedia
Example of Force-Fields¶
Force-fields can be used to measure binding energies. The most commonly used force-fields for macromolecules are Amber, Gromos, and Charmm. Special scoring functions were developed to experimentally reproduce known binding energies. This is discussed in some detail in different parts of this course, in particular in the chapters on molecular docking and molecular dynamics (Free Energy Perturbation method).

Other Methods¶
Other methods that can be used to estimate protein binding affinities also exist. The most accurate approaches are usually more computationally costly. Linear Interaction Energies and Free Energy Perturbation are the most rigorous methods for calculating binding energies. They take advantage of the properties of thermodynamic cycles and aim to predict the free energies of the binding of new ligands from knowledge of the experimental binding energy of a known reference compound.

Incorporation of the Solvent¶
Solvent effects remain a central problem in modeling calculations. Simplified models have been identified to be a major cause of inaccuracy in binding energy calculations. The most rigorous methods consider explicit solvent molecules but they are too slow. Other approaches represent the solvent as a continuum. These issues are discussed in detail in the chapter on molecular dynamics.

Obtaining Macromolecular 3D-Structures¶
Experimental Methods¶
Knowing the exact 3D-structure of bio-molecules is essential for any attempt to understand how these machineries fulfill their function and how they interact with each other. Unfortunately, getting to the structure of such macromolecules in the lab is not easy. X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, and cryo-electron microscopy are the three main techniques that provide protein structures experimentally. The distribution by source of the structures available in the protein data bank is indicated in the diagram below.

X-ray Crystallography¶
X-ray crystallography is a technique in which the pattern generated by the diffraction of X-rays passing through the single crystal lattice of a given material is recorded and then analyzed to reveal the 3D structure of the molecules composing the lattice. X-ray crystallography involves several steps that are presented in the following pages.

wikipedia
Protein Production and Purification¶
The first step is to get large amounts of protein we want to crystallize. This is generally done by cloning the protein in various expression systems such as microorganism, bacteria, plant etc.... The protein is then purified, since only highly purified solutions can give rise to crystals and not every protein can be purified to a sufficient degree.

Growing of Single Crystal¶
The next step is the growth of single crystals. Many approaches are used to produce crystals but the general method is to start with a solution of the protein or its complex and to slowly change the conditions so that saturation is exceeded. This can be done by changing parameters such as ionic strength, pH, temperature of the solution and the addition of seed molecules. Supersaturation has to be reached slowly so that only a few nucleation sites are created which, with time, will grow larger. Thousands of solution conditions may be tried before finding one that succeeds in crystallizing the molecules, and ultimate success is never guaranteed.

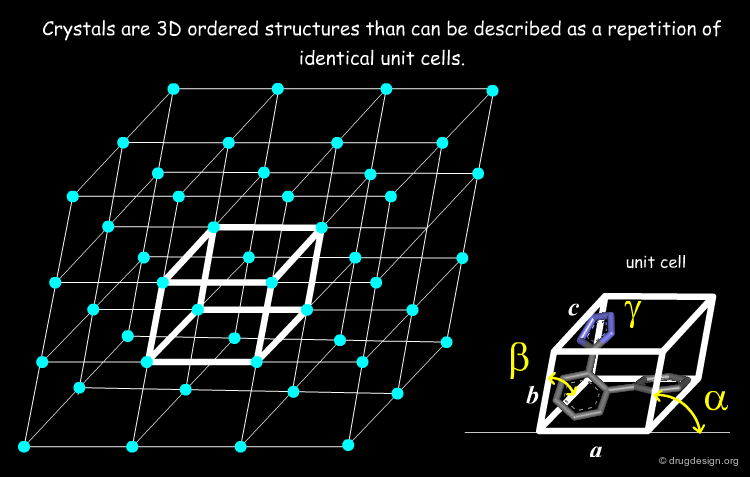
The Single Crystal¶
The starting point of X-rays is a single crystal (or a monocrystal) that must be a crystalline solid in which the crystal lattice of the sample is pure, continuous and unbroken. The basic unit of a crystal is called the unit cell, which is defined by three principal axes (a, b and c) and three angles (alpha, beta and gamma). Single crystals are difficult to obtain in the laboratory. A protein crystal needs to be at least 0.2-0.3 mm in all three dimensions to diffract strongly enough to achieve the resolution needed for a successful 3D structure determination.
Collecting the Diffraction Data¶
The crystal is then bombarded by X-rays, and the interference between rays scattered by different parts of the crystal give rise to diffraction. This diffraction pattern is dependent on the 3D location of the atoms. Diffraction data only record intensity; unfortunately, all information about the phase angle (which relates the various diffraction points and the unit cell) is lost. This is a major hurdle for structure determination.


Recovering the Phase Angle¶
In order to solve the structure the lost phase angle must be recovered. Many strategies have been developed and include multiwavelength anomalous diffraction (MAD), multiple isomorphous replacement (MIR) and molecular replacement methods.

{kind=link}
Structure Determination and Refinement¶
On the basis of the measured intensities of the diffraction points and the recovered phase angles, electron density maps are calculated. The maps are then interpreted and fitted to a 3D structure model. The quality of the structure can be evaluated by its resolution and by comparing the observed diffraction pattern and the one calculated from the final model. On the basis of this comparison the structure can be iteratively refined.

Atomic Coordinates¶
At the end of the process, the 3D structure of the macromolecule is obtained as a list of coordinates (x, y and z). The overall quality of the coordinates is usually measured by two quantities: (1) the resolution; this is the ability to resolve two points separated by some distance from each other. The higher the resolution, the "sharper" our view of the structure. High resolutions of macromolecules are around 2 Å; (2) the crystallographic R factor; it measures the agreement between observed structure factors and those calculated from the model. A well-refined structure of a macromolecule has an R factor of not larger than 15%.

The Advantages of X-ray Crystallography¶
X-ray crystallography has major advantages, making it the most practical method for bio-molecule structure determination. More importantly, it provides accurate atomic resolution models and allows scientists to resolve relatively large structures and complexes. Recent examples are structures of viral capsides and the ribosome, each composed of tens of thousands of atoms.

The Limitations of X-ray Crystallography¶
X-ray crystallography also has some major limitations: (1) the information provides only one snapshot of the protein that does not reflect its dynamic behavior; (2) contacts between molecules in the crystal and the dense packing might affect the structures; (3) the procedure is very slow; (4) there are many macromolecules which can not be resolved, especially proteins with significant hydrophobic portions (membrane proteins).

NMR Spectroscopy¶
Nuclear magnetic resonance (NMR) is a physical phenomenon based upon the magnetic properties of the nuclei of some atoms. NMR spectroscopy was developed in the mid-1980s enabling the elucidation of 3D structures of macromolecules in solution. The first NMR structures deposited at the PDB started to appear in the early 1990s.

Media
This picture was released into the public domain wikipedia
{kind=link}
NMR Concepts¶
NMR spectroscopy takes advantage of the fact that some nuclei have spins. The spinning charges generate a magnetic field that will either align with or against an applied external magnetic field. Absorption of energy supplied by electromagnetic radiation at specific frequencies (resonance) can cause the spin to reverse its orientation. The relaxation process of the spins can help to characterize the system. The most basic contribution to the resonance of a particular nucleus is the local electronic environment. For example electronegative atoms or delocalized electrons in aromatic rings will cause a shift of the resonance frequency in neighboring spin systems ("chemical shifts").

Spin-Spin Coupling¶
The interactions between neighboring spins ("spin-spin coupling") give rise to the line splitting in NMR spectra. These interactions provide connectivity information because they occur only between protons on covalently bonded atoms. Another spin-spin interaction can occur between non-covalently bonded nuclei in space (the Nuclear Overhauser Effect - "NOE"). This interaction is distance dependent and therefore only protons separated by less than 5Å can be observed. This is the most crucial information for structure determination.

Data Collection¶
Since the signal from the NMR experiment is exceedingly weak, repeated scans are necessary to improve the signal-to-noise ratio. Signals arising from the decay of all resonances are measured simultaneously. This complex signal is then deconvoluted by the use of Fourier transforms into a set of frequencies and their corresponding amplitudes. Classical 1D NMR is useless for analyzing the 3D structure of macromolecules (due to the spectral overlap of hundreds of protons) and therefore 2D and 3D NMR techniques are used. Correlated spectroscopy (COSY) gives rise to cross peaks corresponding to the covalent structure (spin-spin couplings) while in NOESY cross peaks are obtained from the NOE effect.

Structure Determination¶
Once all the resonances have been assigned, the outcome is a list of rough inter-proton distances from the intensity of the NOE signal between interacting protons and some dihedral angles from spin-spin coupling constants. 3D models consistent with the set of constraints are then generated. These models (usually several dozen) can be refined by calculating NOEs from the model and fitting against the measured NOE intensities, as well as by optimizing for more favorable structural properties.
Analysis¶
Finally, when an ensemble of refined structures is obtained which satisfies the experimental restraints and has reasonable stereo-chemical parameters, we are ready to analyze the data. From the ensemble one can, for example, calculate deviations in the position of individual atoms, derived from the multiples structures. This information is believed to be related to the dynamics of the structure in solution. The quality of the final structure depends on both the number and quality of the NOE measurements.

The Advantages of NMR¶
The main advantage of NMR spectroscopy over X-ray crystallography is that there is no need to have the sample in a crystalline state, which restricts the applicability of X-ray crystallography. Crystal contacts may also affect the structure. The positions of all atoms, including hydrogens, are obtained. Crystal structures positions of hydrogens can only be determined at very high resolution. Finally, information about the dynamics of the molecule in solution can be deduced.

The Limitations of NMR¶
NMR based approaches for structure determinations have some major limitations. The precision of the technique is still not as high as in X-ray crystallography. In addition, only relatively small bio-molecules (for proteins usually smaller than 300 residues) can be solved. According to the size of the putative proteins (Open Reading Frames - ORFs) encoded in different genomes (see bars in different colors in the figure), less than half of the proteins can be solved by current NMR techniques (the limit is indicated by the dashed red line in the figure). Rapid developments in equipment and software has led however, to continual improvement in these areas.
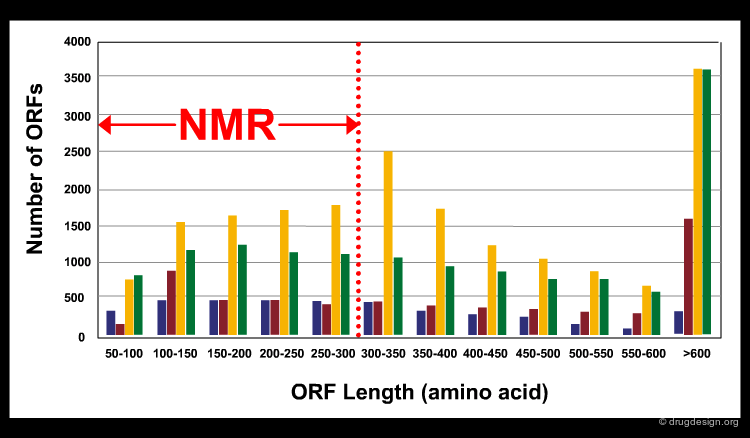
articles
NMR spectroscopy: pushing the limits of sensitivity Spiess HW Angew Chem Int Ed Engl 47 (4) 2008
The structure of the neuropeptide bradykinin bound to the human G-protein coupled receptor bradykinin B2 as determined by solid-state NMR spectroscopy Lopez JJ, Shukla AK, Reinhart C, Schwalbe H, Michel H, Glaubitz C Angew Chem Int Ed Engl 47 (9) 2008
Electron Microscopy¶
Electron microscopy (EM) is another approach that provides information about the 3D structure of macromolecules. It has recently become popular in structural biology as it can provide molecular structural data about large molecular complexes.
wikipedia
Media
This picture was taken from the RCSB PDB website PDB Protein Databank
Basic Concept¶
The electron microscope uses electrons to create an image of the target. It has much higher magnification or resolving power than a regular light microscope, allowing it to see very small objects such as cells and large molecules. The technique currently employed to solve structure, which was the breakthrough in solving bio-molecule structures by EM, is cryo-electron microscopy ("cryo-em"). The images are created with an electron microscope equipped with a "cold stage". This means that the samples are scanned in a frozen state. This procedure protects them from radiation damage and keeps them in a native state.

The Advantages of Electron Microscopy¶
The most important advantage of EM is the ability to resolve very large biological complexes. Sometimes EM is the only alternative. One example is the 3-dimensional atomic model of a protein called tubulin that actively participates in cell division. At a resolution of 3.7 Å EM provided the first highly detailed look at tubulin. Structures determined by EM can serve as an initial reference for interpretation of X-ray diffraction patterns. In addition, structural information about transition states and intermediate states in chemical reactions can be collected.

The Limitations of Electron Microscopy¶
The limitation of EM is the structural resolution that can be achieved. Even though theoretical analysis suggests that there is no problem in achieving high resolutions, technical issues such as optical problems and noise, prevent reaching very high amplifications. For example, the current resolution (about 3.5 Å) is not sufficient to determine the location of the amino-acid side chains.

Copyright © 2024 drugdesign.org