Encoding Molecules¶
Info
In order to be manipulated by computers chemical structures, chemical reactions and their associated properties must be encoded in computer-readable means. The present chapter reviews methods currently used and gives concrete examples to illustrate them.
Number of Pages: 124 (±3 hours read)
Last Modified: May 2009
Prerequisites: None
Encoding Chemical Structures in 2D¶
Encoding Chemical Structures in 2D¶
Chemical structures must be encoded in order to be manipulated with computers. Computer-readable internal representations, chemical file-formats, linear notations and the canonicalization of 2D chemical structures represent different aspects in the encoding of molecules. This is discussed in the following pages.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Internal Representation¶
The internal representation of a 2D chemical structure concerns the topology, the topography and the graph of the molecule. These concepts are described in some detail in the pages that follow.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Topology¶
Topology is the mathematical study of properties of objects which are preserved through deformations, twistings, and stretchings. Molecules built from the same atoms, but with different topology are called constitutional isomers in chemistry.
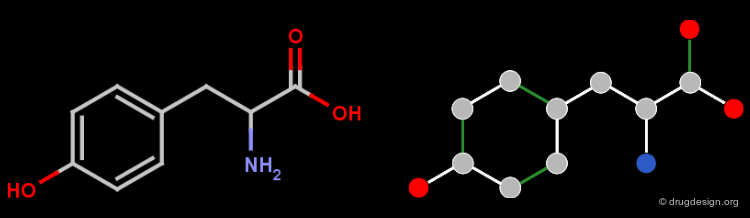
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Topography¶
Topography describes the position of the objects in the three dimensional space. Topography changes through deformations, twistings, and stretchings. Molecules having the same topology but different topography are called conformers in chemistry.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Chemical Graphs¶
A graph is a network of nodes (vertices) that are connected by edges. Chemical structures can be well represented by graphs in computer programs, the atoms are nodes, the edges are bonds. The graph representation provides some important advantages: easy to implement with software programs, the graph theory has solid mathematical background and provides many useful algorithms.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Basic Graph Representations - Adjacency Matrix¶
An adjacency matrix is an nxn matrix, each column and row represents a node. A connection between two nodes is marked with a boolean (yes/no: 1/0) value in the corresponding cell. It is very fast to access the elements of this data structure, effective for dense graphs (many connections) but ineffective for chemical graphs, that are usually sparse as the majority of the cell values are zeros. Extension of the graph with new nodes is slow, because the entire matrix has to be reallocated. The matrix is symmetric to its diagonal, so some implementations save memory by allocating the half matrix only.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Basic Graph Representations - Adjacency List¶
An adjacency list is a list of various length arrays, the index of adjacent nodes (connected neighbors) are listed for each node. The adjacency list is more compact for sparse graphs like chemical structures. New atoms can be added by adding a new array and reallocating the arrays of the adjacents. A connection appears in the redundant adjacency list twice, so some implementations reduce the memory footprint by storing a connection only once.
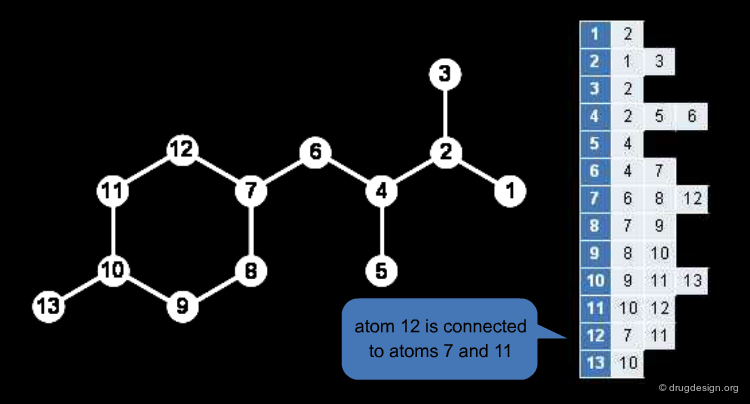
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
The Redundant Connection Table¶
Chemical structures are labelled graphs, nodes (atoms) and edges (bonds) both have important properties. The connection table is a common description format of chemical graphs. Atoms and their properties (atom type, charge, radical, stereo parity) are listed, and the hydrogens are usually omitted.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Markush Patents¶
Eugene A. Markush filed a patent application in the United States in 1923 for a method of preparing pyrazolone dyes. The quite unspecific claim ("homologues of aniline and halogen substitution products of aniline") was granted in 1924. Markush Structures describe a set of molecules with a single structure - a scaffold containing variable ligands. Markush structures frequently appear in patent applications, but they are used to describe combichem libraries as well.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Markush Structures¶
The graphs of Markush structures usually contain R-atoms and their R tables, or other special atoms like generic atoms (hetero, halogen) atom lists (O, S), link nodes (n=1-4), or pseudo atoms (alkyl, alkoxy) often in AND/OR relations. Structure searching and overlap ananalysis of Markush databases is a challenging task as one structure can represent huge or even infinite libraries, so the enumeration of the members is often impossible.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Problems of Graph Representation¶
Problems that occur in graph representation are multiple. Some of them are described in the following pages.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Stereoisomerism¶
Since chemical structures are often represented by two dimensional drawings, stereoisomers are identical graphs. These problems can be solved by the extension of the standard topological graph model with coordinates and stereo properties (i.e. parity).

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Canonicalization¶
A molecule can be drawn in many ways resulting different graphs. Hydrogens are often omitted but not always, and some of them are mobile (tautomerism). Mesomers like nitro group can be represented by resonant forms. Charges can be delocalized and the various representations of salts (even having stoichiometry issues) make the situation more complex.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Macromolecules¶
Many macromolecules can be represented by graphs, but those graphs are often too complex for graph functions. Fortunately, most of the peptides and nucleic acids contain repeating sequences of few block types, thus string-based representations and string operations can be more effective than graph based ones in this field. Polymers usually contain mixtures of molecules with various sizes, which makes graph representation very difficult.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Metalorganics¶
The classic graph model must be extended for coordanitive systems like ferrocenes. The coordinative bonds can be connected more than two atoms in the graph model (hyperedge), while visually connected to one or sometime no atom at all.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Chemical File Formats¶
We present here a second aspect of chemical representation: the chemical file-formats.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Chemical Structure Files¶
The most common chemical structures are molecules. Other structures can be reaction schemes, query structures (substructures often with variable parts) and Markush structures (set of molecules described by a common scaffold and ligand variations). Chemical structure files can be binary or text based. Though the binary files often embed much more than the structure (labels, text boxes, graphics), they are proprietary and application dependent, so they are usually not portable between systems. The text based file formats are widely used they are standards and supported by most chemical applications.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
MDL Chemical Table File Formats¶
MDL (now Symyx) introduced the CTfile family (Chemical Table file) in the 80's which soon became de facto standard for the connection table based description of chemical structures specifying both connectivity and spacial coordinates. The most popular members of the CTfile formats are:

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
The Structure of a Molfile¶
The Molfile of L-phenylalanine is shown in the figure below.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Header Block of a Molfile¶
The header block of the Molfile of L-phenylalanine contains the following information.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Counts Line of a Molfile¶
The counts line of the Molfile of L-phenylalanine contains the following information.
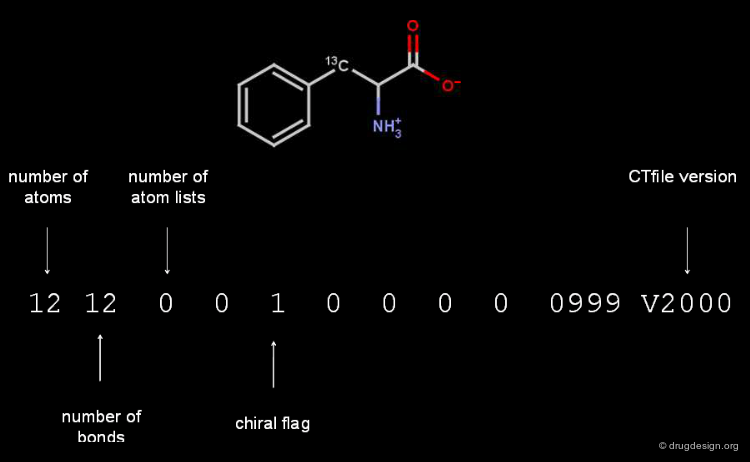
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Atom Block of a Molfile¶
The atom block of the Molfile of L-phenylalanine contains the following information.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Bond Block of a Molfile¶
The bond block of the Molfile of L-phenylalanine contains the following information.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Property Block of a Molfile¶
The property block of the Molfile of L-phenylalanine contains the following information.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SDfile¶
An SDfile contains a list of molecules separated by $$$$ marker lines. Each molfile can be followed by a property block containing named properties.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
The Molfile Format - Summary¶
One important limitation of the Molfile format comes from the fixed array size of the FORTRAN implementation. Originally, no more than 256 atoms were allowed in molfiles and the fixed character position specification did not allow the extension of this limit to more than 999 atoms. In case of druglike molecules, however, this is not a serious limitation. Though MDL introduced new alternatives (V3000 version and the XML based XD format) to overcome the limitations of the V2000 molfile format, it is still one of the most popular chemical structure file formats.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Chemical Markup Language (CML)¶
CML (Chemical Markup Language) is an XML based public standard file format supported by many commercial and open source tools. Similarly to molfiles it represents molecules with a connection table in a very flexible, though space inefficient format. CML allows to assign complex data hierarchy to the structures.
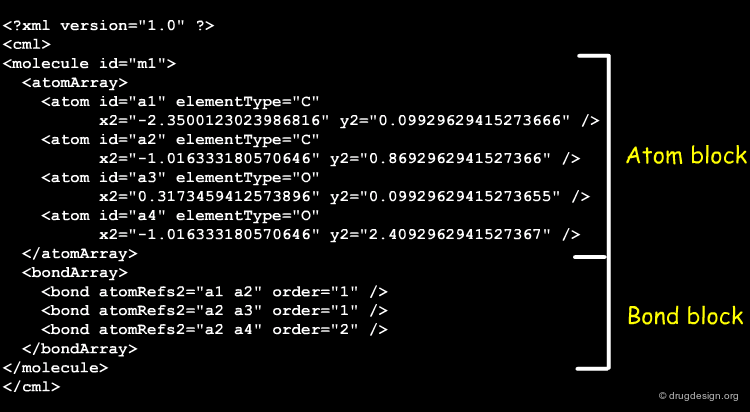
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Molecular Constitution in Linear Notation¶
The third aspect of chemical representation concerns the description of molecular structures in linear notations.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Linear Notations - Chemical Nomenclature¶
Chemists use traditional nomenclature systems to name compounds, describing the positions of ligands attached to a core scaffold. The current IUPAC document specifying the standard naming rules for organic compounds is about 200 pages long. The new provisional document consists of more than 1300 pages and it is still not complete!

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Naming Software Tools - Autonom¶
Autonom (AUTOmatic NOMenclature), the first naming program was developed by Janusz L. Wisniewski and released in 1989 at Beilstein Institute. Autonom supports IUPAC nomenclature in a Beilstein dialect.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
articles
AUTONOM: System for Computer Translation of Structural Diagrams into IUPAC-Compatible Names. 1. General Design. J.L. Wisniewski Journal of Chemical Information and Computer Sciences 30 1990
Other Naming Software Tools¶
Autonom was integrated in the Beilstein Database and was available in MDL applications as well. Modern structure drawing programs are usually free for academics (such as ChemDraw, Marvin, ChemSketch) and provide molecule-to-name and name-to-molecule functions. The batch versions, however, are usually sold separately.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Line Notations¶
The character based description of chemical structures are as old as computers themselves. In the age of character based interfaces chemists had to specify molecules in a single line of text format. The line notations are very compact (sometimes 50 times smaller than Molfiles), and they usually do not store coordinates.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Wiswesser Line Notation¶
Wiswesser Line-Formula Chemical Notation invented by William J. Wiswesser in 1949, was the first line notation capable of precisely describing complex molecules. It is not used nowadays, but it was popular in the 60's and 70's.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
articles
How the WLN began in 1949 and how it might be in 1999 William J. Wiswesser J. Chem. Inf. Comput. Sci. 2 1982
ROSDAL Line Notation¶
The Beilstein Institute worked out the ROSDAL notation (Representation of Organic Structures Description Arranged Linearly) with more focus on the computer parsability than human readibility.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES Line Notation¶
The SMILES notation (Simplified Molecular Input Line Entry Specification) is the most popular line notation today. It is a computer parsable but quite human readable chemical format developed by Dave Weininger in the late 80's at Daylight Chemical Information Systems. The elemental atoms are represented with their symbols in brackets, but the "organic subset" of atoms can appear without the brackets. In this case they are considered to be filled with hydrogens.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES: Isotopes Ions, Radicals¶
Isotopes, charged atoms and radicals are always enclosed in brackets. If an atom symbol is within a bracket, the hydrogens must be written inside.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES: Chains¶
The aliphatic bonds between atoms are indicated with - (single), = (double), # (triple) and : (aromatic) symbols. Single bonds can be ignored between the aliphatic atoms. Branches are enclosed in parentheses. Disconnected structures are separated by periods.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES: Rings¶
Atoms closing a ring are marked with a number. Aromatic atoms are written in non-capital characters, the default bond type between aromatic atoms is the aromatic bond. To avoid ambiguity, the hydrogens must be written on aromatic nitrogen atoms.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES: Stereo¶
The relative cis-trans stereo configuration of double bonds is encoded with / and (slash, backslash) symbols. The relative tetrahedral stereo configuration is indicated by @ (clockwise), and @@ (anti-clockwise) characters, that must appear in the brackets of the center atom.
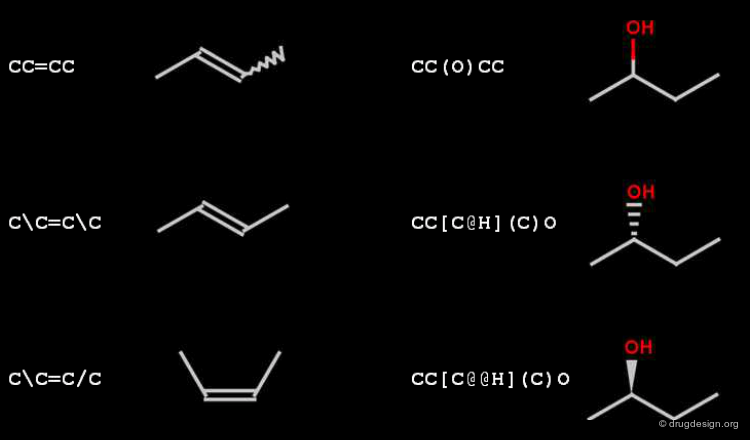
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES: Reactions¶
SMILES supports reactions as well, the reaction arrow is simply indicated by >> (double chevrons). Molecules written between the two chevrons are considered agents.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
SMILES: Summary¶
The SMILES format is compact (less database and memory space), easily parsable by computer programs, and the structural characteristics are usually visible for the experienced eye. As the SMILES format does not contain whitespace, additional data fields can be appended to the SMILES string separated by tabs or spaces. Several extension make SMILES one of the most versatile formats of chemoinformatics. SMARTS for example - a query extension of SMILES - is probably the most powerful chemical query language.
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
InChI¶
InChI (IUPAC International Chemical Identifier) is linear notation format supported by IUPAC to produce canonical strings from the chemical structures. It disconnects salts and organometallic compounds, and normalizes protonation and charge. It then applies the classical Morgan algorithm for sorting the atoms in canonical order (see the next chapter for details). Specific parts of the InChI string are called layers.
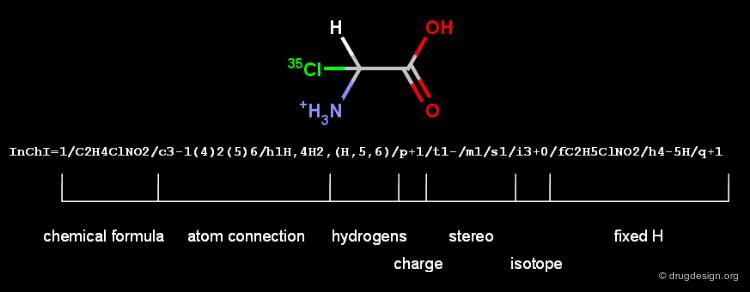
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
InChI - Standard by a Single Source¶
InChI is a developing standard, supported by IUPAC, and this support makes it an important format. It is freely available, but not freely modifiable. The canonical form is not based on a standard method, but provided by a shared single source code. It is not permitted to write an InChI generator program, the original source code must be used without modification. Since it is written in C, adding InChI functionalities to managed memory applications (like Java or C# ) is not easy.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Canonicalization¶
Canonicalization of molecule files is an effort to represent a chemical entity with a unique and unambiguous text string. Such a string could be used to identify the molecule in every document or database by a simple text search, but a canoncial string is not suitable for substructure searching.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Canonicalization of Molecules¶
Canonicalization is not file format dependent, canonical InChI, IUPAC name, SMILES, SLN string would be equally good (Molfile as well, but it is multiline and not too compact).

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
The Canonicalization Progress¶
A good canonicalization method produces the same unique string from any representations of the molecule, and it never provides the same string for different molecules. Additionally, the molecule structure should be reconstructed from the string itself, that makes the canonical molecule string different from any unique identifier numbers.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Chemical Standardization¶
The canoncialization progress starts with a chemical standardization to convert mesomers, tautomers, salts and other structural features to a standard format. This makes the chemical graph canonical.
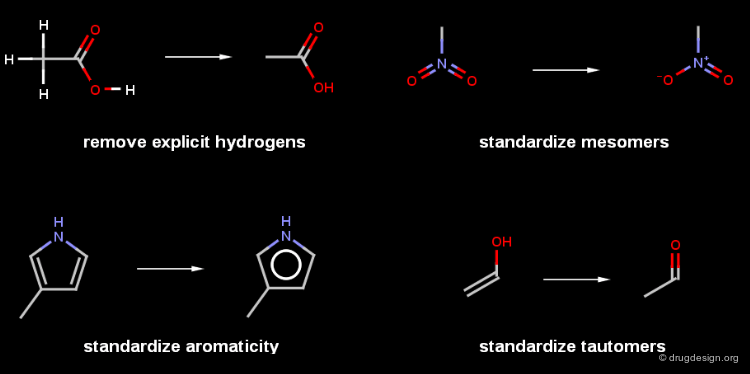
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Standardization of Salts¶
There is no universal standard for the representation of structures, that is true with more emphasis for salts and organometallic compounds. The representations of these compounds vary implementation by implementation. The salts are often converted to ionic form, solvent molecules like water or ethanol appearing with the molecules are usually removed.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Normalization of Atom Order¶
When the chemical graph is standardized, the order of atoms must be normalized to produce a unique text string independently from the drawing order of the atoms (connection tables usually contain atoms in the drawing order).
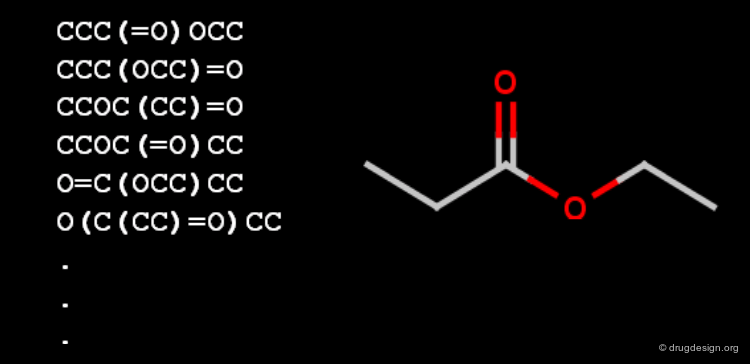
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Canonicalization with the Morgan Algorithm¶
The most popular sorting method is the Morgan algorithm. It consists of a two-step procedure: (1) labeling and (2) the calculation of invariants.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
The Morgan Algorithm: Labeling¶
The Morgan algorithm starts with assigning a label to all nodes of the graph. It first labels the nodes with their number of connections (degree of the graph node), then it adds the labels of the adjacent atoms to each atom label. The loop repeats until the number of different labels is not increasing any further.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
articles
The Generation of a Unique Machine Description for Chemical Structures - A Technique Developed at Chemical Abstracts Service H. L. Morgan J. Chem. Doc. 5 1965
The Morgan Algorithm: Calculating Invariants¶
The topologies of chemical structures are often symmetrical, but the labelling algorithm can include the atomic numbers, the bond order, charges, isotopes and many other chemical information to resolve symmetry issues. When all labels are different, each atom gets an index called invariant.
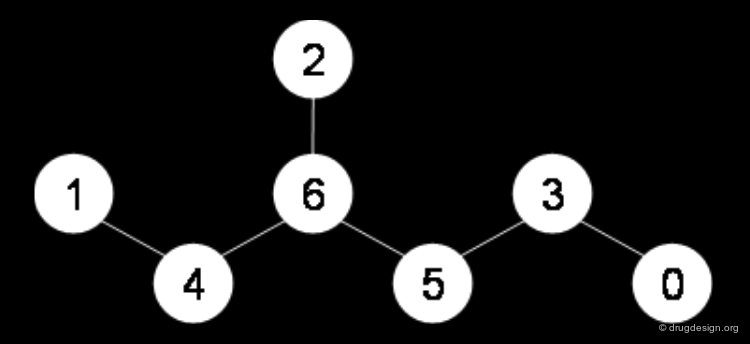
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Creating the Canonical File¶
The canonical file can be written then in any formats using the atom order of the graph invariants which is independent from the drawing order.
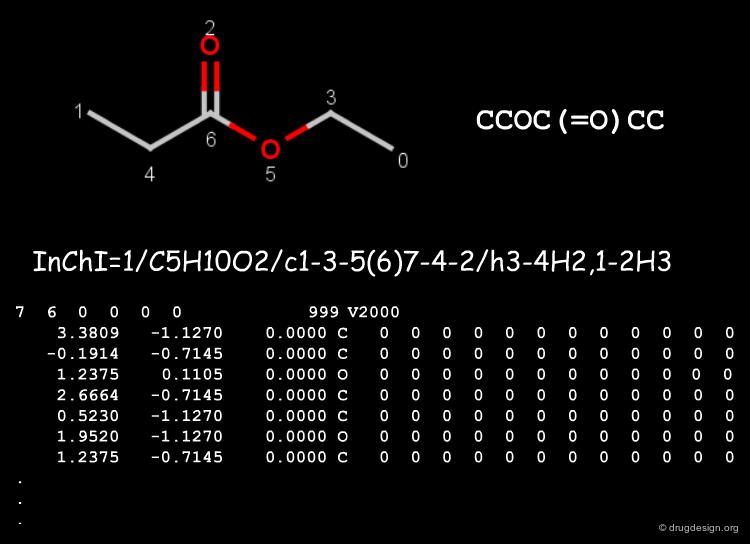
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Graph Invariants and Symmetry¶
The graph invariants are very useful to symmetries in molecular structures. The beautiful stereochemical variance of chemical structures makes the canonical file generation very difficult in some cases. The most of the tools fail to generate canonical files in such cases.

Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Encoding Stereochemistry¶
If a molecule is described in 3D, the encoding of its stereochemical centers is straightforward because the stereochemical information is embedded in the 3D coordinates. On the contrary if the molecule is described by its 2D connectivity, no stereochemical information is given in these diagrams and the file-format must be therefore extended to include stereo parity information, as explained in the following pages.

General Solutions¶
When chemical structures are described in 2D (connectivity) or in 1D (as in SMILES) the stereochemical information is described in a way allowing to rapidly store, search, compare and retrieve molecules or reactions. Current programs use atom-centered or bond-based chirality specifications, as illustrated in the file-formats SD, SMILES and CML, in the following pages.
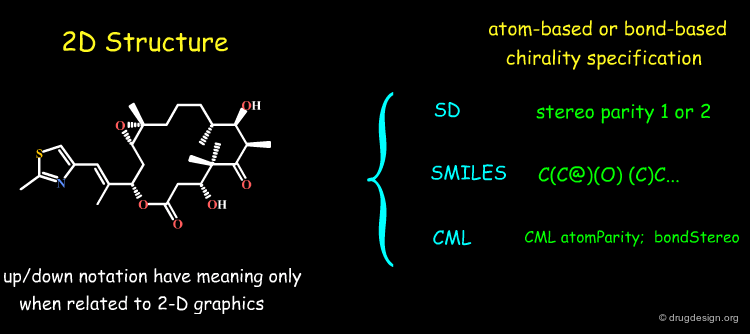
Coding Stereochemistry in SD Files¶
In SD files (MDL format) an atom-based chirality is used to specify the chirality: 0 = no stereo; 1 = odd; 2 = even and 3 = unspecified). The stereo parity is assigned as explained on the second page.

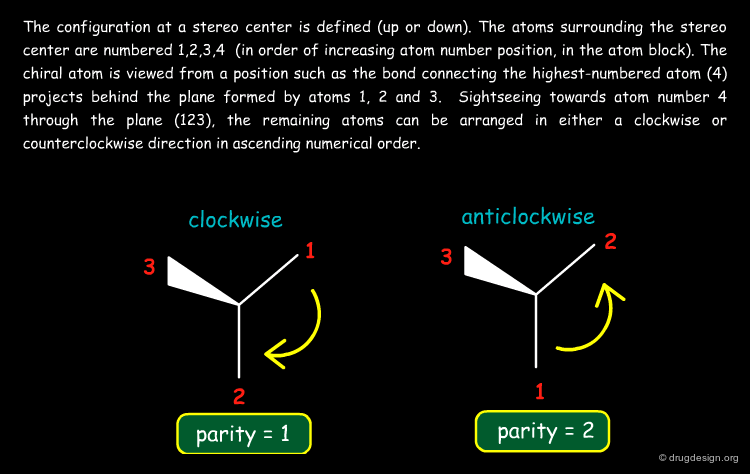
Author
Gyorgy Pirok Chief Technology Officer, ChemAxon, Budapest, Hungary
Stereochemistry in SMILES¶
In SMILES chirality marking is defined by using the atom's left-to-right order (in the SMILES string). A chiral center can be written starting from any of its neighbor atoms, and the choice of whether to list the remaining neighbor in clockwise or anticlockwise order is also arbitrary. The following SMILES representations are all equivalent and specify the same chiral center displayed in the view.

Stereochemistry in CML¶
CML (Chemical Markup Language) describes the components of chemical structures in a language that is perfectly well understood by computers and not intended to medicinal chemists for "manual" translation into their own 2D representations. CML is flexible and powerful. Global, local and complex stereochemistries (e.g. metal coordination) can be described in a number of ways. Below is given a short extract of CML describing stereochemistries.
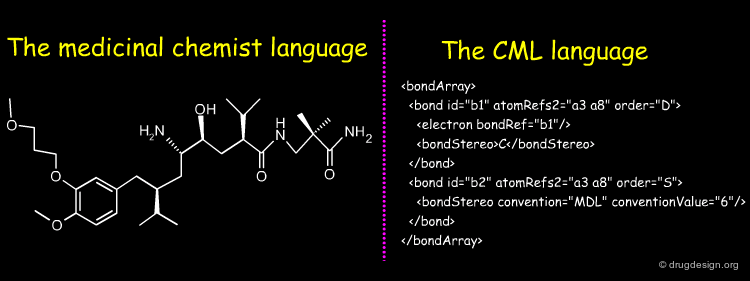
articles
Chemical Markup, XML and the World--Wide Web. 4. CML Schema P. Murray-Rust and H. S. Rzepa J. Chem. Inf. Comput. Sci 43 2003
The Internet as a Chemical Information Tool H. S. Rzepa, P. Murray-Rust and B. J. Whitaker Chem. Soc. Revs
1997
Chemical Markup, XML, and the Worldwide Web. 1. Basic Principles P. Murray-Rust and H. S. Rzepa J. Chem. Inf. Comput. Sci 39 1999
Chemical Markup, XML and the World--Wide Web. 2. Information Objects and the CMLDOM P. Murray-Rust and H. S. Rzepa J. Chem. Inf. Comput. Sci 41 2001
Chemical Markup, XML, and the World-Wide Web. 3. Toward a Signed Semantic Chemical Web of Trust G. V. Gkoutos, P. Murray-Rust, S. Rzepa and M. Wright J. Chem. Inf. Comput. Sci 41 2001
Development of Chemical Markup Language (CML) as a System for Handling Complex Chemical Content P. Murray-Rust, H. S. Rzepa and M. Wright New J. Chem
2001
Encoding 3D Structures¶
Encoding 3D Structures¶
A 3D structure is characterized by a molecular architecture that needs to be analyzed, searched, visualized, compared with other structures and exchanged with other groups. As the practice of manipulating 3D molecular structures with computers has matured, specific methods for encoding 3D structures were adopted by the scientific community that are presented in this section.

3D File Formats¶
The intensive use by many groups of the Protein Data Bank has triggered the adoption of the format proposed by this organization for encoding the 3D structures of molecules. This supplanted many other formats such as those used in force-field programs or those derived from simple 2D formats. We present here some of the most commonly used file-formats used for 3D structures.

PDB Introduction¶
The PDB was established at Brookhaven National Laboratories in 1971 as an archive for biological macromolecular crystal structures. Later structures from other sources were included. The Protein Data Bank was transferred in 1998 to the Research Collaboratory for Structural Bioinformatics (RCSB) in San Diego. The PDB database is of central importance and used by a broad number of scientists whose work is related with the 3D structure of macromolecules.
PDB Example¶
The PDB files primarily hold information about structure. However, additional details must be given. This includes many technical details about the experiments, indications about the quality of the data and other biological relevant information. PDB files are usually very big. The atomic coordinates taking the vast majority of the space. The additional information is located before the coordinates in the header section of the file. Moreover the PDB site provides a number of tools enabling to search, visualize and analyze macromolecular 2D and 3D data.

PDB Fields Explained¶
The coordinate lines of a PDB file start with field name "ATOM" for atoms of macromolecule (protein/nucleic acids) or "HEATATM" for atoms of other molecules such as ligands, ions or solvent. Following the details about the identity of each atom (its type, the amino/nucleic acid, its index number) appear the three Cartesian coordinates (x, y, z) in units of angstroms that describe the position of the atom in space.

CIF¶
The intensive use of the information derived from crystallographic structures of small molecules has triggered the development more extensive normalization formats to describe large amounts of crystallographic that can be parsed by computer. The CIF (Crystallographic Information File) - a "meta data" representation, was introduced in the late 1980s. The following is an extract of CIF file.
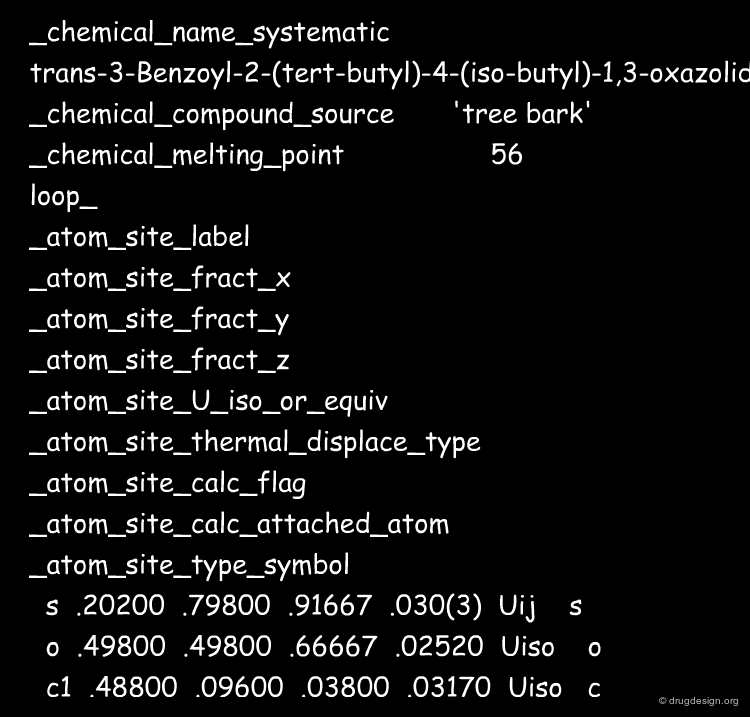
book
Book Series International Tables for Crystallography International Tables for Crystallography Volume G: Definition and exchange of crystallographic data Springer Netherlands 2005
mmCIF¶
In the mid 1990s the CIF representation was extended 3D macromolecular information and led to the mmCIF (macromolecular Crystallographic Information File) representation. Massive information can be treated, compared and searched. Moreover a "mmCIF dictionnary" enables to check mmCIF data. An extract of a mmCIF file is shown below.

Z-Matrix¶
The Z-matrix is a way to represent the conformation of a molecule in internal coordinates (bond lengths, bond angles, and torsional angles). This is way of representing molecules is often used in quantum mechanical programs, in particular one can follow more easily the changes in geometry optimization. Z-matrices can be easily converted to cartesian coordinates. Below is illustrated the Z-matrix for ethylamine.

3D File-Format Converters¶
The great diversity of programs with 3D molecular structures as input and output, led to development of 3D file format converters. Babel is an example of such converter. It can read, write and convert over 80 different chemical file formats.

Encoding Molecular Surfaces¶
Encoding Molecular Surfaces¶
Along the years, general standards have been adopted for the visualization of molecules and molecular surfaces with their associated coloring scheme. We present in the following pages some of these standards.

Ball and Stick¶
Here the molecule is displayed as the assembly of atoms and bonds. Atoms are represented as small spheres and bonds as tubes.

Van der Waals - CPK Surface¶
In this type of representation (also called space filling or CPK) the molecule is defined as a set of spheres of van der Walls radii of the individual atoms.

Connolly-Surface¶
The Connolly surface is somewhat similar to the van der Waals, however it is mathematically defined in order to make it smoother. The Connolly surface is defined as the contact surface and the inward facing part of a probe sphere when it is simultaneously in contact with more than one atom.
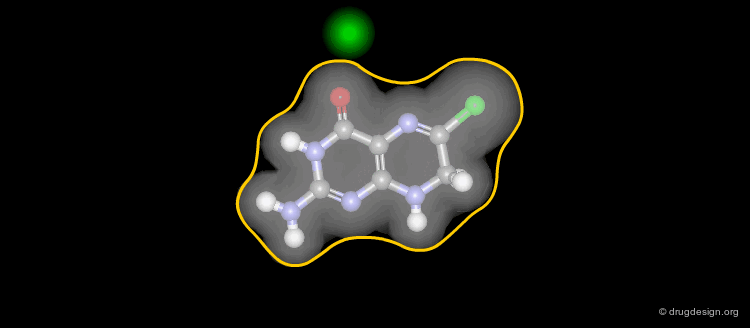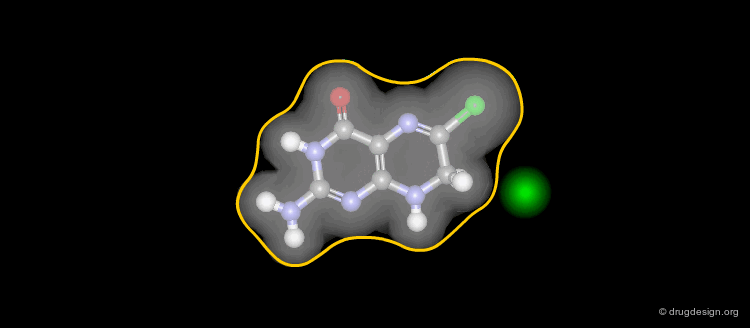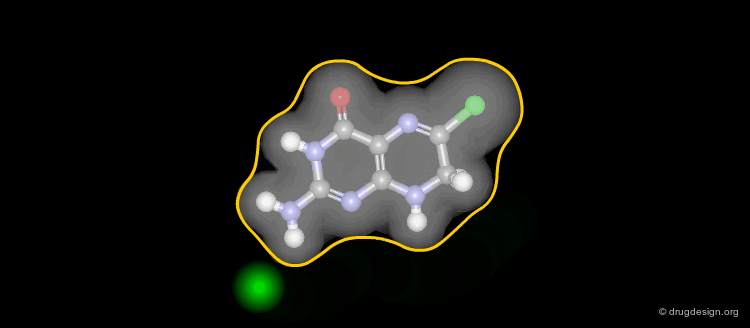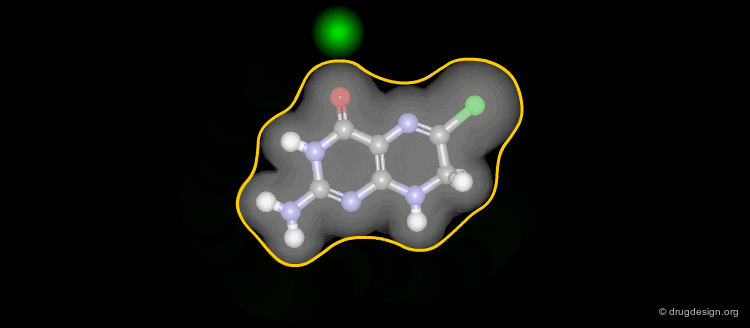

Solvent-Accessible Surface¶
The solvent accessible surface corresponds to the molecular envelope of the surface of each atom which is accessible to the center of a probe sphere of a given radius, generally 1.4 Å (sphere including a water molecule).


3D Isopotential Contour Lines¶
In cartography contour lines are drawn in a plane. They can also be drawn on the surface of a 3D object: for example on the 3D envelope of a molecule. Here for example, the contour line connects points with the same property value (e.g. electrostatic potential) on the surface-accessible surface and the number associated to the curve, indicates the value of this property. With the advent of colored computer graphic displays this plotter-based method has been supplanted by the 3D color-coded real time visualization of surface properties.

articles
3-D Molecular Lipophilicity Potential Profiles, A new Tool in Molecular Modeling P. Furet, A. Sele, N.C. Cohen Journal of Molecular Graphics 6 1988
book
N.C. Cohen ACS Symposium Series volume 112; Symposium on Computer Assisted Drug Design; American Chemical Society, Washington,DC. 1979
Coloring by ...¶
There are many ways of coloring a view. In the following pages we present some of the most typical ways of coloring 3D representations: by atom-type, by molecule, by color, by properties.

By Atom-type¶
In this representation the atoms are colored according to the atomic color-code defined and a color is also given to half of the bonds of this atom.

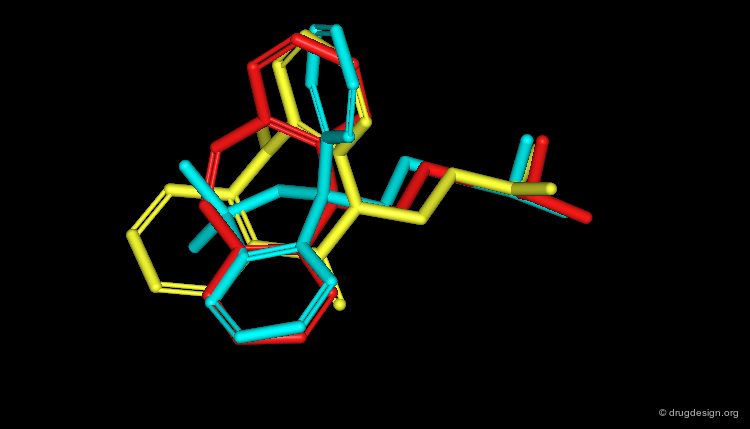
By Molecule¶
A different color is given to each of the molecules visualized in the screen. It is useful when several molecules are superimposed to distinguish and recognize the individual molecules in the view.
By Color¶
All or part of a given molecule is visualized in one color. Interactive "picking" of the molecule or a set of atoms allows one to associate a color with a specified set.

By Properties¶
Molecular surfaces can be color-coded, which adds an entirely new dimension in the understanding of the molecular properties. In the view below the color-coded electrostatic potential created on the surface of a molecule is displayed (negative=red, positive=blue, neutral=white).

Encoding Reactions¶
Encoding Reactions¶
A chemical reaction is characterized by its reactants, products and reaction conditions, but has no information on the mechanism of the reaction. A chemical mechanism, on the other hand, describes the electron movements, reaction rates and stereochemistry involved. Thus, representing a chemical reaction is not necessarily the same as representing a chemical mechanism. Representing reactions is also somewhat synonymous with classifying reactions, due to the fact that in order to classify something, it must first be represented.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
book
Willett P Proceedings of a conference organised by the Chemical Structure Association University of York, July 1985 Gower Publishing Company Limited 1986
Schleyer et al
J Wiley and Sons 1998
J Gasteiger Handbook of Cheminformatics Wiley-VCH 2003
Describing a Chemical Reaction¶
For the SN2 reaction the list of atom and bond changes during the reaction is as follows: C-Br bond broken (bond order 1 -> 0); C-Cl bond formed (bond order 0 -> 1); Br atom charge change from 0 -> -1; Cl atom charge change from -1 ->0. Any reaction which undergoes the same set of atom and bond changes would be considered part of the same generic reaction. BUT this does not describe the mechanism.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Representing Reactions¶
There are several methods for representing chemical reactions, in part due to the closed source nature of much of chemistry, and many reaction representation are tied to specific pieces of software. In the table below are listed some of the strategies used for representing reactions.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Extension of Molecular File Formats¶
One strategy is based on the extension of existing molecular file formats. In the following pages we present those based on SMILES, SMIRKS and RXN.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Daylight's SMILES™ Molecular File Format¶
SMILES™(Simplified Molecular Input Line Entry System) allows molecules and reactions to be specified using ASCII characters representing atom and bond symbols. This format can also fully handle stereochemistry.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Daylight's SMIRKS™ Molecular File Format¶
SMIRKS™ (SMILES ReaKtion Specification) is a superset of SMILES™ used for expressing reactions in the general form: REACTANTS > AGENTS > PRODUCTS as illustrated below, in the acid catalysed esterification reaction.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
SMILES and SMIRKS: in Conclusion¶
The advantages and disadvantages of the SMILES™ and SMIRKS™ file formats for representing chemical reactions are listed below. "Canonicalizable" means that it can be transformed into a unique string to be used as a universal identifier for a specific molecule or reaction.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
The RXN Reaction File Format¶
Created by MDL (now part of Symyx) the RXN file format is an extension of the MOL file format and has capability of mapping, but only heavy atoms. It is one of the most widely used formats - most, if not all, chemical software can handle RXN files, allowing for the interconversion of other file formats with RXN. However, it does not allow for the description of reversible reactions, mapping of proton movements or non-linear reactions, i.e. it can only handle A + B --> C + D.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
The RXN File Explained¶
The RXN file format is essentially a list of molecules involved in the reaction.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
The Molecule¶
The molecule portion of the RXN file is made up of two "blocks" the atom block, and the bond block. Each of these is identical to the format found in the MOL and SDF file formats.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
The Atom Block¶
The made up of lines of the format is shown below: atomic coordinates, atom symbol, mass difference, charge, atom stereo parity, hydrogen count etc...

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
The Bond Block¶
Atom numbering in MOL files is implicit, the first atom listed is atom 1, second atom 2, etc. The bond block is made up of lines of the format: first atom in the bond, second atom in the bond, bond type, stereo, double bonds (cis or trans), bond topology, reacting centre status ...

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Graph Approaches to Representing Reactions¶
Graph based approaches, whilst not reaction file formats, represent a valuable method of representing reactions on computers. Two of them will be discussed here: the Arens and the Vladutz models.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Arens Representation of Reactions¶
Arens (1979) derived a classification system for all reactions in which the reaction is considered as a cyclic point system. Any indication of the nature of the atoms or their substituents is omitted. Only those bonds that are between the reacting points are considered and the bond orders are represented by the symbols 3 for triple, 2 for double, 1 for single and 0 for no bond.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
articles
** ** JF Arens Recl. Trav. Chim. Pays-Bas 98 1979
** ** JF Arens Recl. Trav. Chim. Pays-Bas 98 1979
** ** JF Arens Recl. Trav. Chim. Pays-Bas 98 1979
Vladutz Representation of Reactions¶
Vladutz represented reactions where the key atoms of the reactants and products are displayed only once. Giving a multigraph with labeled vertices (atoms) and edges (bonds) that makes use of different edge labels for those edges that are either not affected or altered by the reaction (formed → or cleaved -/-). These "superimposed reaction graphs" (SRG) can be further reduced by omitting any vertices that do not participate in any altered edge, which give the "superimposed reaction skeleton graphs" (SRSG).
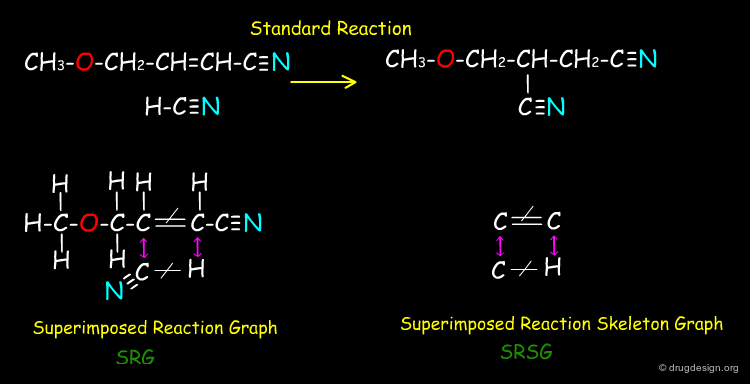
Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Matrix Representation of Reactions¶
Matrix based approaches also do not represent a file format, but it is a useful method of representing reactions on computers. Only one method will be discussed here: the Dugundji-Ugi Model.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
The Dugundji-Ugi Model¶
The theory of "be"- (bond and electron) and "R"- (reaction) matrices is an algebraic model of the logical structure of constitutional chemistry and is commonly known as the Dugundji-Ugi Model. It is based upon the statement shown below. The be-matrices represent constitutional formulas, which in turn represent the graphs of the chemical constitution of molecules. They do not represent only single molecules and are genuine mathematical constructs.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
articles
An Algebraic Model of Constitutional Chemistry as a Basis for Chemical Computer Programs Dugundji J. and Ugi I. Top. Curr. Chem. 39 1973
Computer-Assisted Solution of Chemical Problems - The Historical Development and the Present State of the Art of a New Discipline of Chemistry Ivar Ugi, Johannes Bauer, Klemens Bley, Alf Dengler , Andreas Dietz , Eric Fontain, Bernhard Gruber, Rainer Herges, Michael Knauer, Klaus Reitsam , Natalie Stein Angwe. Chem. Int. Ed. 32 1993
Models, Concepts, Theories, and Formal Languages in Chemistry and Their Use as a Basis for Computer Assistance in Chemistry Ivar Ugi, Johannes Bauer, Carola Blomberger, Josef Brandt, Andreas Dietz, Eric Fontain, Bernhard Gruber, Annette v. Scholley-Pfab, Antje Senff, and Natalie Stein J. Chem. Inf. Comput. Sci. 34 1994
Example of Dugundji-Ugi Model¶
The molecular assembly of n atoms is described by its symmetric nxn be-matrix. The ith row (and column) is assigned to the atom Ai(1 {#8804} i {#8804} n). The entry bij (= bji, i {#8800} j) of the ith row and jth column is the formal bond order of the covalent bond between atoms Ai and Aj. The diagonal entry bii is the number of free valence electrons at the atom Ai.The redistribution of valence electrons by which the reactants of a chemical reaction are converted to their products is represented by the R-matrices. The fundamental B + R = E equation of the Dugundji-Ugi model corresponds to the additive transformation of the be-matrices B of the reactants by the reaction matrix R into the be-matrix E of the products.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
articles
An Algebraic Model of Constitutional Chemistry as a Basis for Chemical Computer Programs Dugundji J. and Ugi I. Top. Curr. Chem. 39 1973
Representing Reactions using Relational Databases¶
Relational databases can also be used to represent reactions; a good example of this is the KEGG (Kyoto Encyclopedia of Genes and Genomes) Database. Here reactions are textually represented e.g. the reaction is defined both textually (with the Definition field) and as a series of compound names (Equation) and then graphically, which comes from the compound names being linked to MOL files.

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
CMLReact: an XML Vocabulary for Chemical Reactions¶
CMLReact (Chemical Markup Language for Reactions) describes a set of components which add chemical reactions to CML (Chemical Markup Language). The design of CMLReact was guided by all the methods previously mentioned, as well as the need to represent mechanism, map atoms and bonds and represent non-linear reactions. The CMLReact Schema includes XML constructions for: reactant, product, spectator (e.g. protein, nucleic acid) or substance (which may include solvent, catalysts, etc), mechanism and transition state, annotations (e.g. titles, names and labels), properties (controlling conditions, observations, measurements), atoms, bonds and electrons. These can be explicitly included with a range of attributes (position, annotation, etc.).

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
articles
Chemical Markup, XML and the Worldwide Web. Part 6. CMLReact; An XML Vocabulary for Chemical Reactions GL Holliday, P Murray-Rust, HS Rzepa J. Chem. Inf. Model. 46 2006
Chemical Markup, XML and the World--Wide Web. 4. CML Schema P. Murray-Rust and H. S. Rzepa J. Chem. Inf. Comput. Sci 43 2003
The Internet as a Chemical Information Tool H. S. Rzepa, P. Murray-Rust and B. J. Whitaker Chem. Soc. Revs
1997
Chemical Markup, XML, and the Worldwide Web. 1. Basic Principles P. Murray-Rust and H. S. Rzepa J. Chem. Inf. Comput. Sci 39 1999
Chemical Markup, XML and the World--Wide Web. 2. Information Objects and the CMLDOM P. Murray-Rust and H. S. Rzepa J. Chem. Inf. Comput. Sci 41 2001
Chemical Markup, XML, and the World-Wide Web. 3. Toward a Signed Semantic Chemical Web of Trust G. V. Gkoutos, P. Murray-Rust, S. Rzepa and M. Wright J. Chem. Inf. Comput. Sci 41 2001
Development of Chemical Markup Language (CML) as a System for Handling Complex Chemical Content P. Murray-Rust, H. S. Rzepa and M. Wright New J. Chem
2001
Simple Example of a Linear Reaction in CMLReact¶
All molecules would be represented as shown for the formic acid molecule shown here. The full representation has been shortened here for ease. Molecules may also represented by references to an external collection of molecules.


Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
A More Complex Example in CMLReact¶
CMLReact can also be used to represent a "scheme" showing two alternative pathways for a chemical process. An overall reactionScheme consists of two subsidiary reactionSchemes. Here it is not appropriate to use reactionStepLists and the individual order of reactions is not completely defined. Each reaction would contain the same components as "r1", i.e. reactantList, reactants, productList and products.
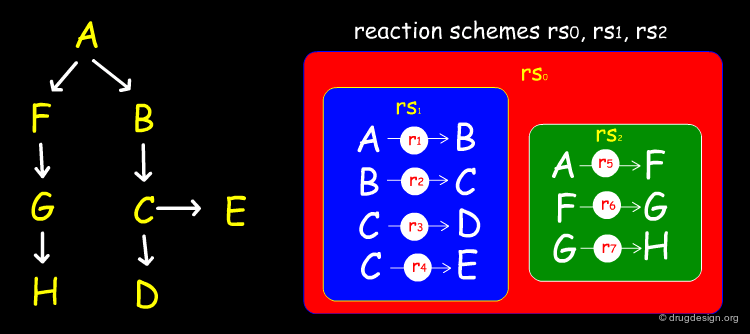

Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
Branched and Cyclic Reactions in CMLReact¶
The type attribute allows us to assign reversibility. Another commonly used attribute is the role attribute, which allows us to assign a specific step in a reaction as being, for example, the rate determining step. Below is shown an example that is translated in CMLReact vocabulary (see second page). For more detailed information on CMLReact see the Holliday article (2006).


Author
Gemma Holliday Research Associate, EMBL-European Bioinformatics Institute, Hinxton, Cambridge, UK
articles
Chemical Markup, XML and the Worldwide Web. Part 6. CMLReact; An XML Vocabulary for Chemical Reactions GL Holliday, P Murray-Rust, HS Rzepa J. Chem. Inf. Model. 46 2006
Encoding Spectral and Analytical Chemical Data¶
Encoding Spectral and Analytical Chemical Data¶
Extensive effort was made to encode in a rational way analytical chemistry data such as molecular spectrometric data, chromatographic data. In the following pages we mention three such systems that were developed on the basis of XML standards; namely: JCAMP-DX, CMLSpect and AniML.

JCAMP-DX: Encoding Molecular Spectrometry Data¶
The Joint Committee on Atomic and Molecular Physics (JCAMP) and the Analytical Instrument Association promoted the development of a format for the storage and interchange of analytical chemistry results. This format named JCAMP-DX enables data interchange for IR/FTIR, NMR, IMS, EPR/ESR, chromatography, mass spectrometry etc...

articles
JCAMP-DX: A Standard Form for Exchange of Infrared Spectra in Computer Readable Form McDonald RS, Wilks PA Appl Spectrosc 42 1988
JCAMP-DX for NMR Davies AN, Lampen P Appl Spectrosc 47 1993
JCAMP-DX for Electron Magnetic Resonance (IUPAC Recommendations 2006) R. Cammack, Y. Fann, R.J. Lancashire, J.P. Maher, P.S. McIntyre and R. Morse Pure and Applied Chemistry 78(03) 2006
JCAMP-DX: A Standard Form for Exchange of Infrared Spectra in Computer Readable Form R. S. McDonald and Paul A. Wilks Appl. Spectrosc. 1 1988
IUPAC/JCAMP-DX An International Standard for the Exchange of Ion Mobility Spectrometry Data J. I. Baumbach, P. Lampen, A.N. Davies Pure Appl. Chem. 73(11) 2001
book
A. N. Davies and H. Hilling and M. Linscheid Software-Development in Chemistry Springer-Verlag, Berlin 1990
CMLSpect: Encoding Spectral and Analytical Data¶
CMLSpect (CML Spectra) consists of a Chemical Markup Language (CML) adapted for managing spectral and other analytical data. It has been designed to be flexible enough to contain a wide variety of spectral data.

articles
Chemical Markup, XML, and the World Wide Web. 7. CMLSpect, an XML Vocabulary for Spectral Data Kuhn, S.; Helmus, T.; Lancashire, R. J.; Murray-Rust, P.; Rzepa, H. S.; Steinbeck, C.; Willighagen, E. L. J. Chem. Inf. Model. 47(6) 2007
AniML: Encoding Analytical Chemistry Data¶
AniML (Analytical information markup language) was developed on the basis of XML standard for analytical chemistry data and consists of a generic data container that permits the storage of arbitrary data. It can be seen as a new way to store and exchange spectroscopy and chromatography data.
Encoding Molecular Properties with Descriptors¶
Encoding Molecular Properties with Descriptors¶
Molecular descriptors are of outmost importance in cheminformatics because they express molecules in a numerical manner. Moreover, they contain in a simple representation the complexity carried by the 2D or the 3D representations of molecules. In the following section we introduce how molecules can be encoded with the use of descriptors.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Molecular Descriptors¶
Numerical descriptors express molecular structures in a numerical representation. They allow us to perform computations with molecular structures for similarity search, diversity analysis and also to develop quantitative predictive models.
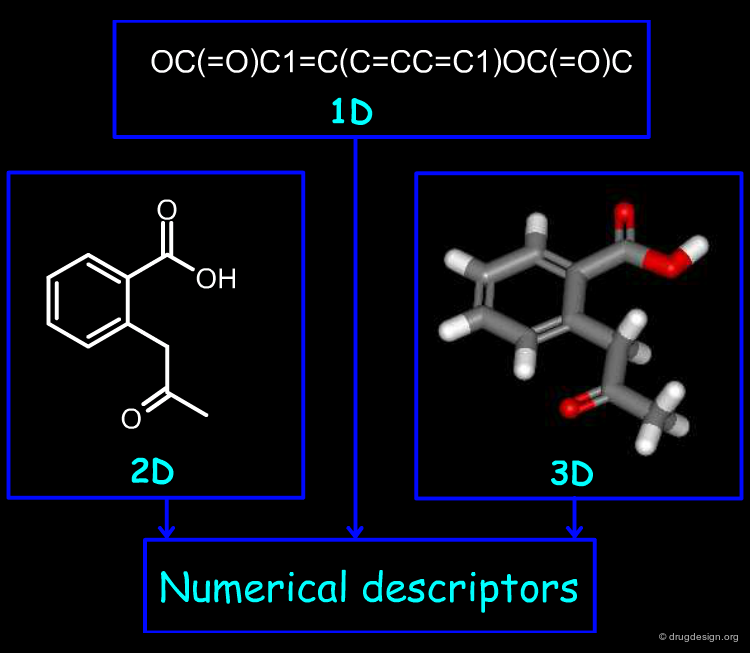
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Types of Molecular Descriptors¶
There are different types of molecular descriptors. They can be geometric, electronic, constitutional, topological or a combination of them. Below are indicated the main characteristics of these descriptors.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Constitutional Descriptors of All Kinds¶
Constitutional descriptors count the occurrence of a feature (e.g. atoms, bonds, chemical groups etc.). They have two important advantages: (1) they are very fast to calculate and (2) they do not need 2D or 3D structures. Some examples of constitutional descriptors are described below.
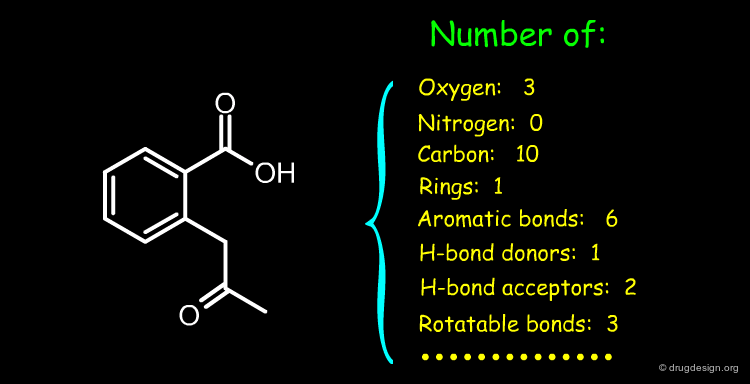
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Topological Descriptors¶
Topological descriptors are generated on the basis of the 2D structure of the molecule. Many descriptors can be derived from the 2D connectivity; in the page that follows we present the Wiener index which is one of the earliest topological descriptors used to characterize entire molecules.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Wiener Index¶
The Wiener index is a topological descriptor characterizing the compactness (or the molecular branching) of a molecule. It just looks at the connectivity of the molecule and ignores atom types. In the formula below, n is the number of atoms in the molecule and dij is the topological distance (number of bonds) between two atoms.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
articles
Structural Determination of Paraffin Boiling Points H.J. Wiener J. Am. Chem. Soc. 69 1947
Recent Advances on the Role of Topological Indices in Drug Discovery Research E. Estrada and U. Uriarte. Current Med Chem. 8 2001
Wiener Index Calculated for 2-Methyl Butane¶
To calculate the Wiener index of a molecule, one has to consider the hydrogen depleted graph of the molecule. The topological distance matrix can be easily constructed, and enable the calculation of the Wiener index.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Variation of the Wiener Index in Analog Structures¶
If we consider acyclic alcanes of formula C6H14 with different branching schemes, we can calculate the Wiener index for each of the isomers and observe that the lower the Wiener index, the more compact the molecule.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Geometrical Descriptors¶
Geometrical descriptors derived from a 3D molecular structure take the spatial arrangement of the atoms in the molecule into account. These can be either elementary descriptors (distances, angles, torsion angles), or global ones such as surface areas, molecular volumes, radius of giration, radial distribution functions, feature trees etc. In the pages that follow we give some details on radial distribution functions and feature trees.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Radial Distribution Functions¶
Radial distribution functions characterize the distribution of an atomic property over a molecule. It provides information regarding bond distances, ring types, planarity or non-planarity. Their calculation require 3D coordinates, though a topological variant can be computed. It is useful for computer-assisted structure elucidation.
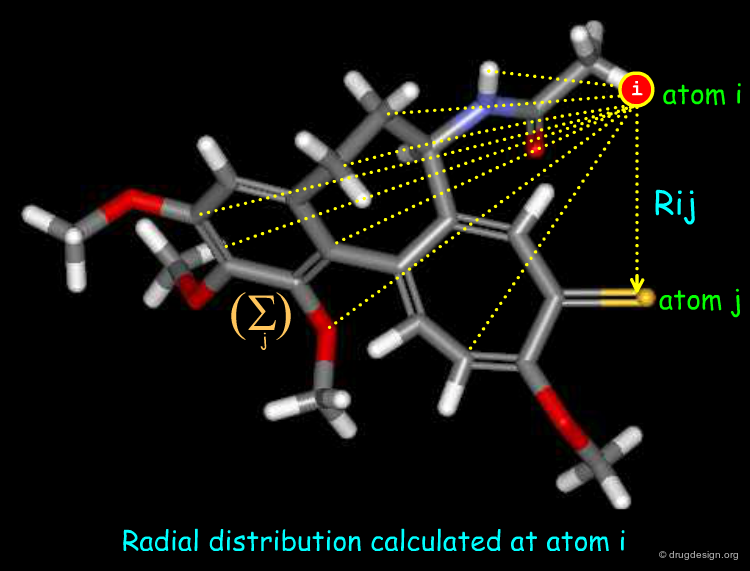
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
articles
The Prediction of the 3D Structure of Organic Molecules from Their Infrared Spectra M. C. Hemmer, V. Steinhauer, J. Gasteiger Vibrational Spectroscopy 19 1999
Calculation of Radial Distribution Functions¶
The general formula of a radial distribution function is shown below. It can be used to calculate the descriptor at different lags. e.g. 1 atom separation, 2 atom separation, 3 atom separation and so on (different R values). A smoothing factor may be used to define the probability distribution of a property.
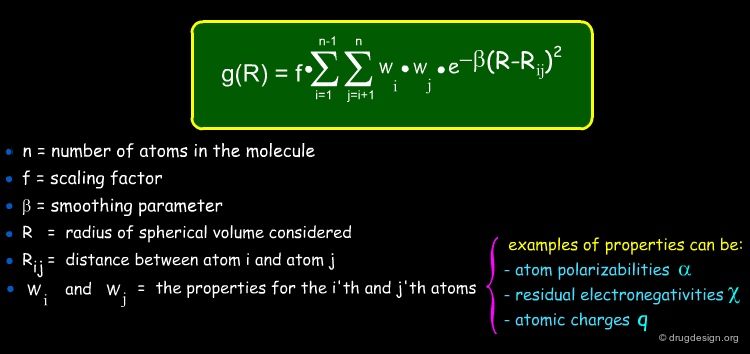
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
articles
The Prediction of the 3D Structure of Organic Molecules from Their Infrared Spectra M. C. Hemmer, V. Steinhauer, J. Gasteiger Vibrational Spectroscopy 19 1999
Feature Trees¶
A feature tree is a topological descriptor representing the molecule as a reduced graph. It generally preserves the arrangement of functional groups and allows molecules to be aligned. Being a 2D descriptor, it is independent of the conformations of the molecules. This descriptor is of great utility for scaffold hopping, virtual screening and the design of combinatorial libraries.
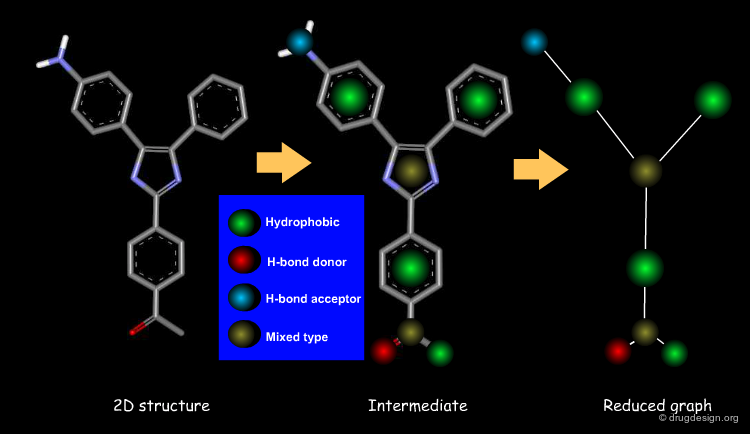
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
articles
Feature Trees: A new molecular similarity measure based on tree matching. Rarey, M. and Dixon, J. S. Journal of Computer-Aided Molecular Design 12 1998
Information Contained in a Feature Tree¶
Below is illustrated an example of molecule with its associated feature tree. In the figure are listed what type of information is contained in a feature tree and how this information is used.
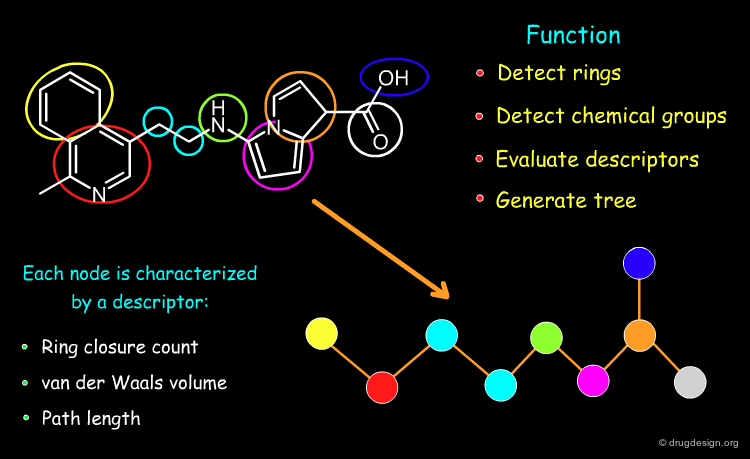
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Feature Trees in Similarity Searching¶
Feature trees are useful fo 2D structural matching. Two trees can be compared in terms of individual nodes, sub-trees or whole feature trees. The algorithms used for similarity searching include: recursive division, match-search and dynamic match-search. Below is illustrated examples of hits found with this method.
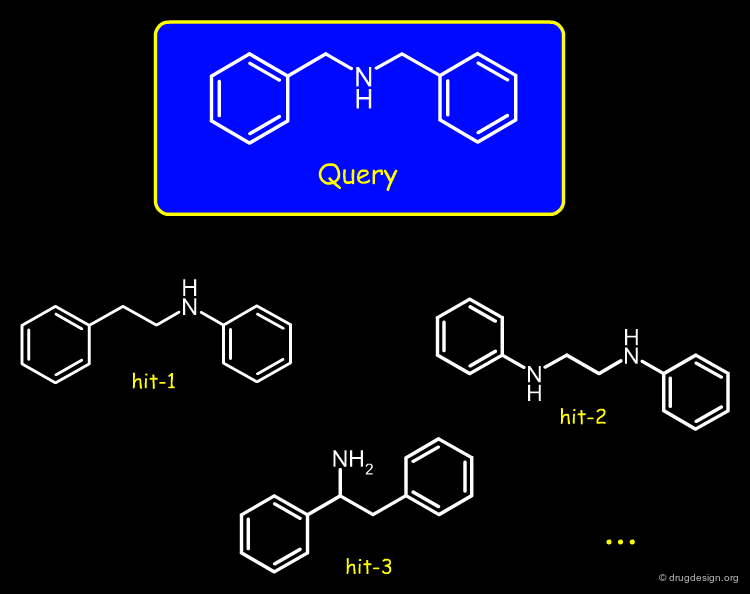
Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Physical Property Calculation¶
Many physical properties can be derived from the 2D or 3D representation of a molecule. In the pages that follow we give some details on the calculation of atomic charges, based on the 2D structure of a molecule.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Gasteiger-Marsili Charges¶
Partial atomic charges are governed by the orbital electronegativity of the atoms. The Gasteiger-Marsili method is an empirical approach to calculate partial atomic charges based on the topology of a molecule. The method is very fast and consists on an iterative procedure that distributes seed charges over atoms, based on orbital electronegativities. Below is shown the Mulliken equation on which the method is based: it relates the orbital electronegativity of an atom to its ionization potential and electron affinity.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
articles
Iterative Partial Equalization Of Orbital Electronegativity- a Rapid Access to Atomic Charges J. Gasteiger, M. Marsili Tetrahedron 36 1980
A New Model for Calculating Atomic Charges in Molecules J. Gasteiger, M. Marsili Tetrahedron Lett.
1978
Electronegativity Equalization: Application and Parametrization W. J. Mortier, K. Van Genechten, J. Gasteiger J. Amer. Chem. Soc. 107 1985
Electronegativity Equalization¶
The process of electronegativity equalization is visualized for methyl-fluorine. The final charges are obtained after 6 iterations, when convergence is achieved.

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Example of Gasteiger-Marsili Charges¶
Initially conceived only for Σ electrons, the Gasteiger-Marsili method has been extended to π systems. An example of charges calculated with the Gasteiger-Marsilli method for methyl benzoate is shown below (for clarity the hydrogen atoms are omitted).

Author
Rajarshi Guha Visiting Assistant Professor, Indiana University School of Informatics, USA
Copyright © 2024 drugdesign.org