Examples of Pharmacophores¶
Info
The present chapter presents how pharmacophores can be derived from a set of known active and inactive compounds. For each example the entire process is described in detail. In some cases the successful exploitation of the pharmacophore is illustrated with designed molecules conforming to the requirements of the pharmacophore.
Number of Pages: 41 (±1 hours read)
Last Modified: January 2004
Prerequisites: Principles in Pharmacophore Elucidation{newLine} Ligand-Based Approaches
ACE Inhibitors¶
Therapeutic Utility of ACE Inhibitors¶
The angiotensin-converting enzyme (ACE) plays a central role in the control of blood pressure through various effects of angiotensin II, which is a potent pressor peptide. ACE inhibitors have the potential to specifically block the formation of angiotensin II and thus reduce high blood pressure.

The ACE Enzyme¶
ACE is a carboxypeptidase enzyme that catalyzes the cleavage of two amino acids from the C-terminal of the Angiotensin I substrate, generating the octapeptide Angiotensin II. Its active site contains a Zinc atom. ACE inhibitors are useful antihypertensive agents.

Discovery of the First ACE Inhibitor¶
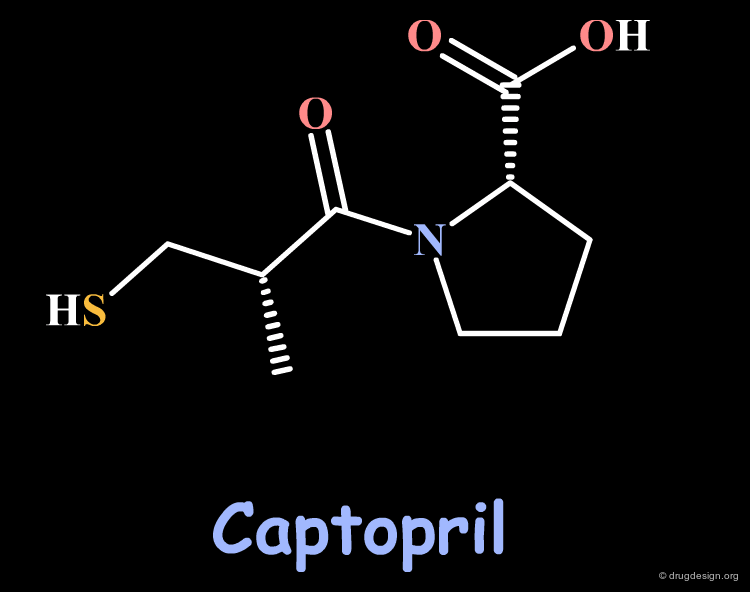
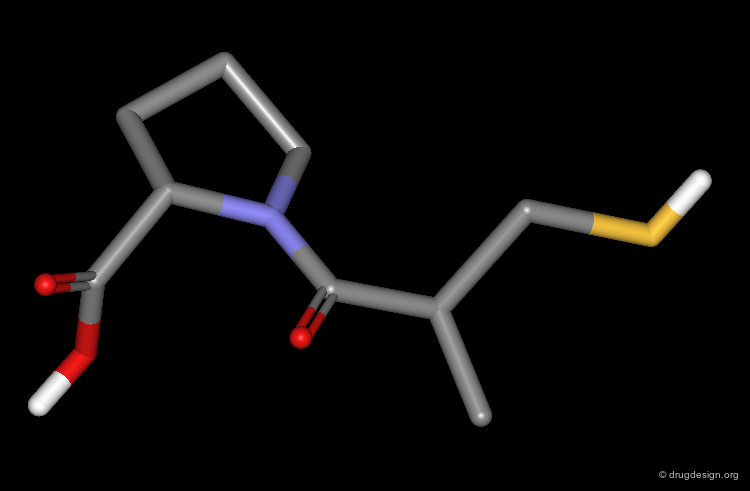
Captopril is the first ACE-inhibitor that was discovered. The validity of the therapeutic concept has been proven by the successful commercialization of this compound whose creation opened an entirely new class of orally active antihypertensive agents.
articles
Design of Potent Competitive Inhibitors of Angiotensin-Converting Enzyme. Carboxyalkanoyl and Mercaptoalkanoyl Amino Acids Cushman DW, Cheung HS, Sabo EF, Ondetti MA Biochemistry 16 1977
Design of Specific Inhibitors of Angiotensin-Converting Enzyme: New Class of Orally Active Antihypertensive Agents Ondetti MA, Rubin B and Cushman DW Science 196 1977
Design of New ACE Inhibitors¶
Based on the structure of captopril, new analogs were designed. Compounds such as the indoline and the tetrahydropyridazine derivatives displayed below are examples of molecules with antihypertensive activities that are more potent than captopril.


Pharmacophore for ACE Inhibition¶
The three basic structural requirements essential for ACE inhibition are: a terminal carboxylate group, a carbonyl group involved in hydrogen bonding, and a moiety such as a SH group, that can bind to a Zinc atom (of the enzyme).


articles
Constrained Search of Conformational Hyperspace Dammkoehler R, Karaswk S, Berkley Shands EF and Marshall G J. Comp. Aided Mol. Des. 3 1989
A Unique Geometry of the Active Site of Angiotensin Converting Enzyme Consistent With Structure-Activity Studies Mayer D, Naylor C, Motoc I and Marshall G J. Comp. Aided Mol. Des. 1 1987
Browser for ACE Inhibition¶

Serotonin Antagonists¶
Therapeutic Utility of Serotonin Antagonists¶
It is suggested that antagonists at the serotonin receptor would have desirable therapeutic potential as anxiolytic agents. Conformational study and superimposition of 4 serotonin receptor ligands (R-methiothepin, spiperone, S-propranolol and buspirone) reveal that they share a common pharmacophore.


Superimposition of Serotonin Receptor Antagonists¶
Superimposition of the amino and aromatic moieties of methiothepin, propranolol, spiperone and buspirone.

Pharmacophore for Serotonin Antagonists¶
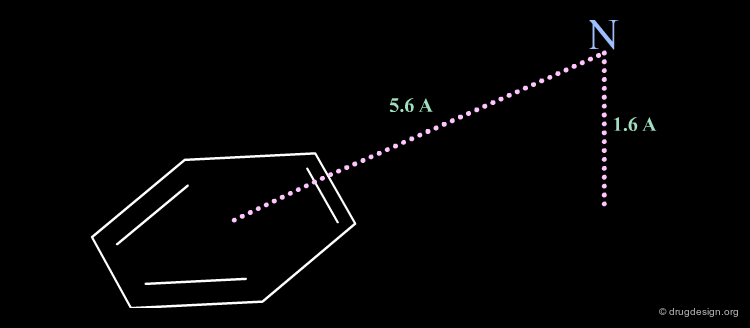
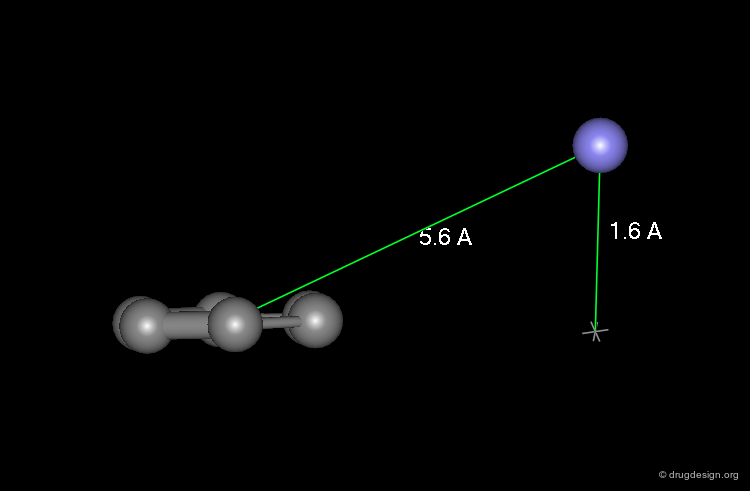
The minimum pharmacophoric elements of serotonin ligand receptors consist of a phenyl ring and a nitrogen atom. The nitrogen atom is located at a distance of 5.6 Å from the center of the phenyl ring and a height of 1.6 Å from the plane of the aromatic moiety.
The Design of MDL 72832¶
The pharmacophore model was used to design new serotonin receptor ligands. MDL 72832 is an example of a designed compound, which showed a nanomolar affinity for the serotonin receptor in vivo.


Browser of Serotonin Receptor Antagonists¶

Dopamine D-1 Agonists¶
Therapeutic Utility of Dopamine D1 Agonists¶
Dopamine receptors have been primary targets for drugs used in the treatment of central nervous system disorders, such as Parkinson's disease or schizophrenia. The following set of dopamine agonists was considered for the development of a model for dopamine D1 receptor recognition.


Pharmacophore for Dopamine D1 Agonists¶
The 3D superimposition of the different dopamine receptor agonists indicated in the previous screen reveals the following common 3D structural features: an amino group, a phenyl moiety and two possible hydroxyl groups at distances as indicated in the figure.


Browser of Dopamine Receptor D1 Agonists¶

Dopamine D-2 Antagonists¶
Utility of Dopamine D2 Receptor Antagonists¶
Psychosis is a thought disorder characterized by disturbances of reality and perception. It is well established that all clinically effective antipsychotic agents act by modulating central dopaminergic activity. This action is interpreted by the ability of these substances to act as antagonists at dopamine receptors in the CNS (central nervous system). At least five distinct dopamine receptors (D1 to D5) have been characterized; however it appears that the specific interaction of antipsychotic drugs with the D2 receptor is most important to their therapeutic action.
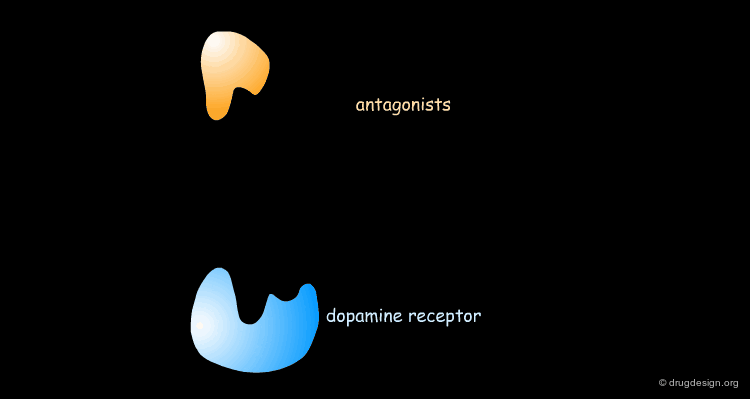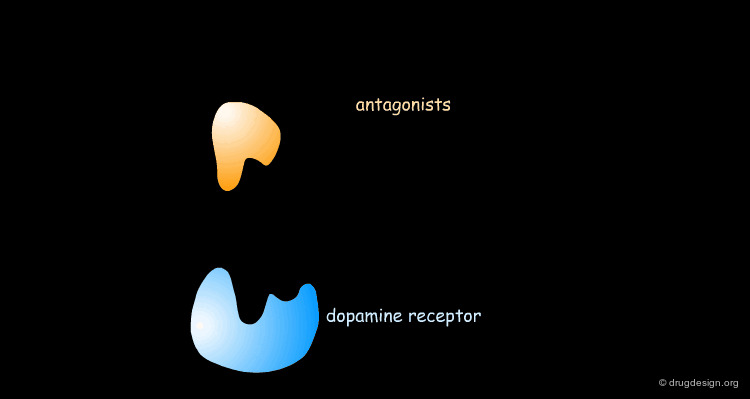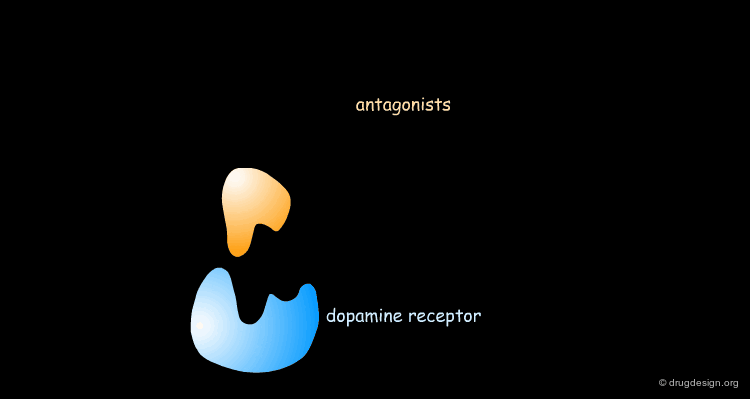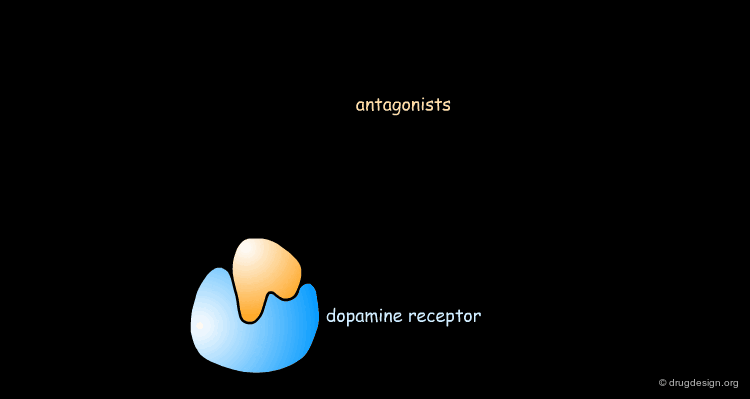
Some Antipsychotic Agents¶
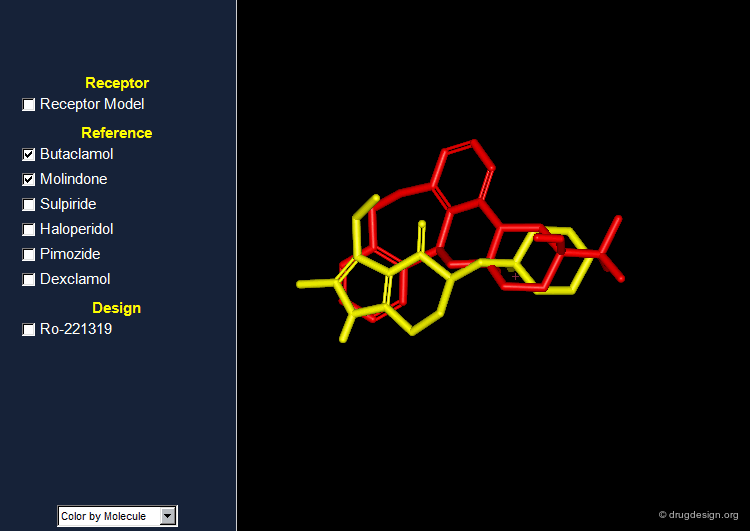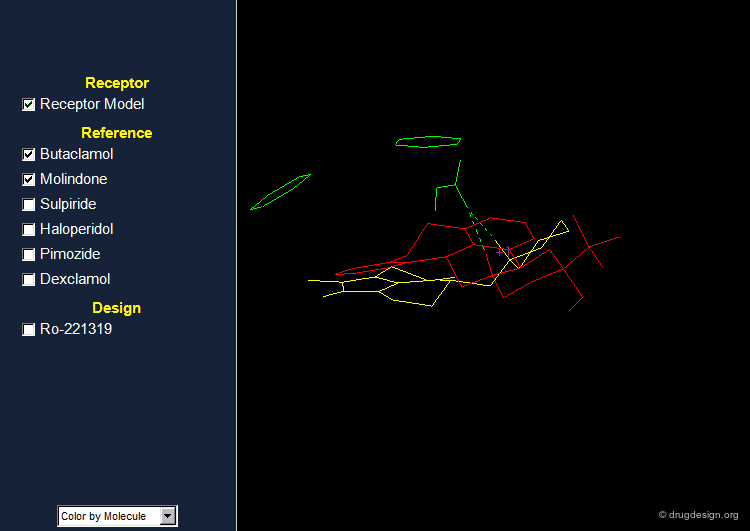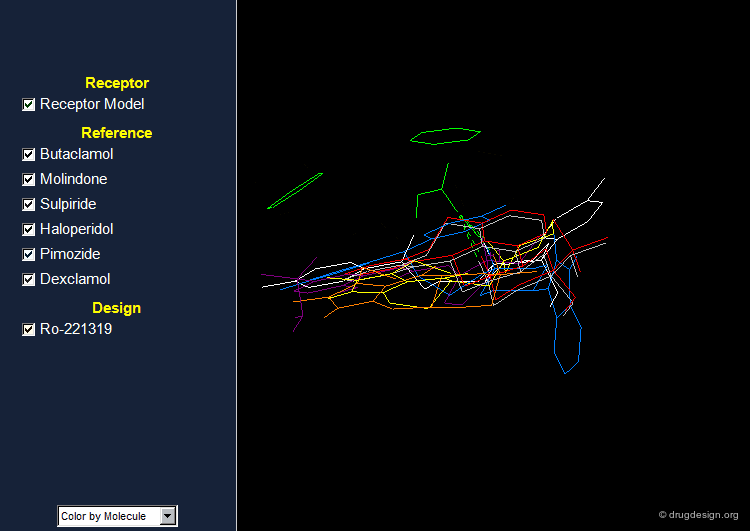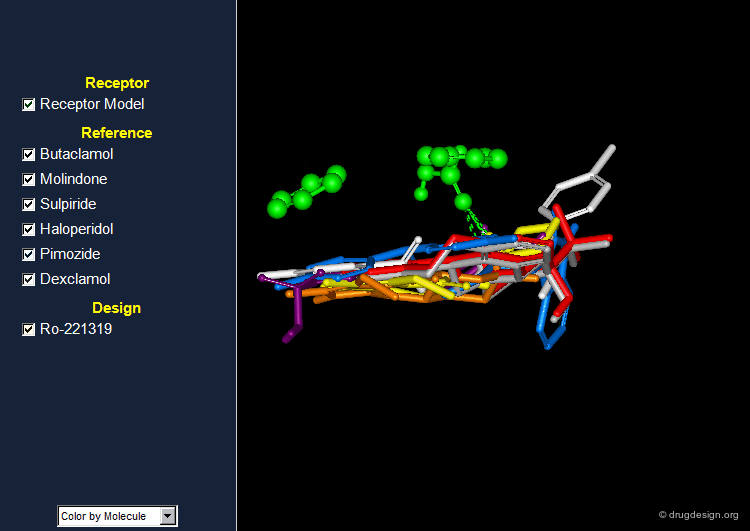
Butaclamol and molindone are examples of antipsychotic agents interacting with the dopamine D2 receptor.
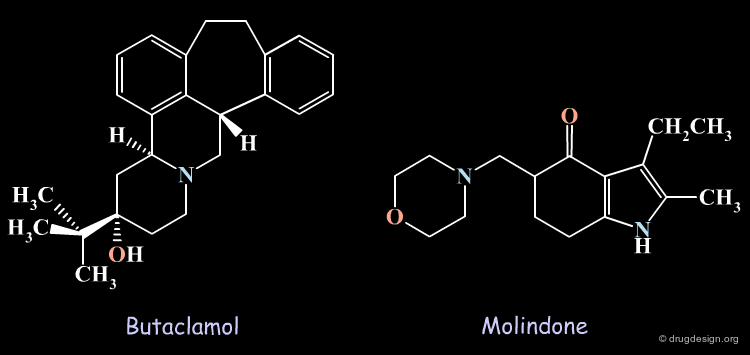

Reference Set of D2 Receptor Antagonists¶
Several other dopamine D2 receptor antagonists were also discovered and attempts to identify common pharmacophoric elements were made. The rigid 3D structure of butaclamol has greatly facilitated the identification of the putative bioactive conformations of the different antipsychotic agents and the way they interact with the dopamine D2 receptor.

Pharmacophore for Dopamine D2 Antagonists¶
Based on the conformations of the various antipsychotic agents shown on the previous screen, a model of the central dopamine receptor was developed and consists of the following: A carboxylate group which could interact with an amino group of the ligand.

articles
The Incorporation of Butyrophenones and Related Compounds into a Pharmacophore for Dopamine D2 Antagonists Froimowitz M and Cody V Drug Des. Discov. 15 1997
Molecular Modeling of the Dopamine D2 and Serotonin 5-HT1A Receptor Binding Modes of the Enantiomers of 5-OMe-BPAT Homan EJ, Wikstrom HV and Grol CJ Bioorg. Med. Chem. 7 1999
Dopamine D2 Receptor Model Explains Binding Affinity of Neuroleptics: Piquindone and its Structure Activity Relationships Teeter MM and Durand CJ Drug Des. Discov. 13 1996
Stacking Interactions¶
A phenyl group Pi-1 which could form a stacking interaction with the ligand's aromatic moiety

Second Aromatic Ring¶
A second aromatic binding site Pi-2 which could interact with a phenyl ring or a ketone carbonyl permitting p-p or n-p ligand-receptor interactions.

The Design of RO-221319¶
On the basis of the chemical frame of molindone and subsequent to extensive analyses of the stereochemical requirements of the dopamine receptor model, a potent antipsychotic agent, Piquindone Ro-221319 having a pyrroloisoquinoline moiety, was designed.


Browser of D2 Receptor Antagonists¶
Aldose Reductase Inhibitors¶
Therapeutic Utility of Aldose Reductase Inhibitors¶
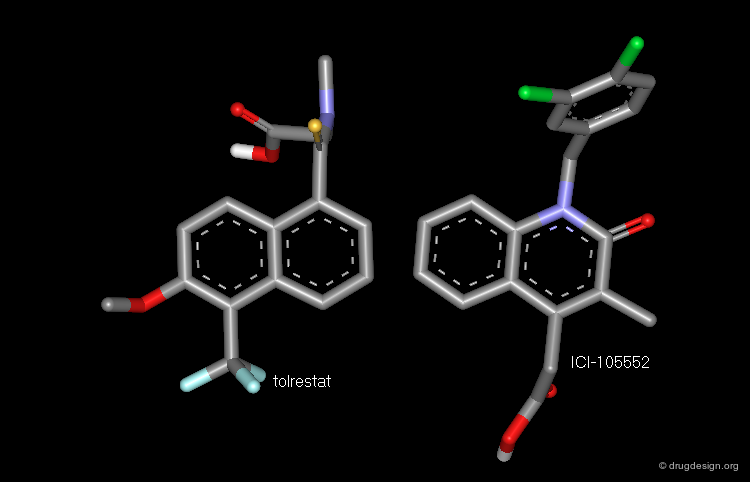
Aldose reductase is the first enzyme in the sorbitol pathway. Inhibition of aldose reductase is a potentially useful strategy for the treatment of diabetic disorders. Tolrestat and ICI-105,552 are examples of potent aldose reductase inhibitors.

Pharmacophore for Aldose Reductase Inhibitors¶
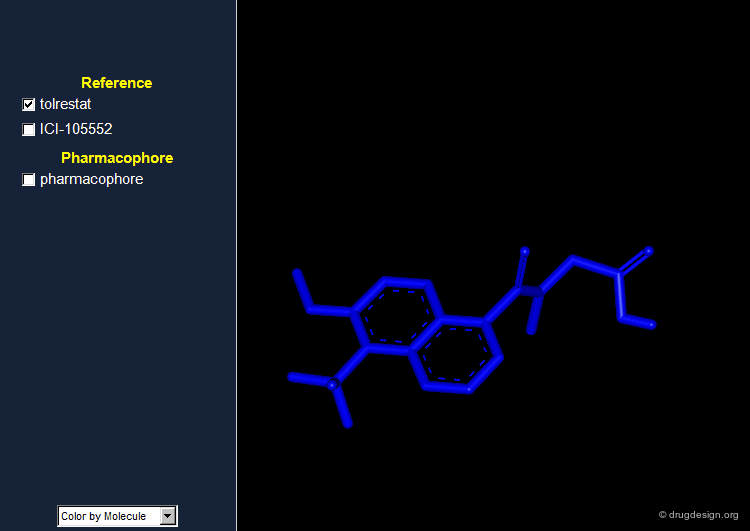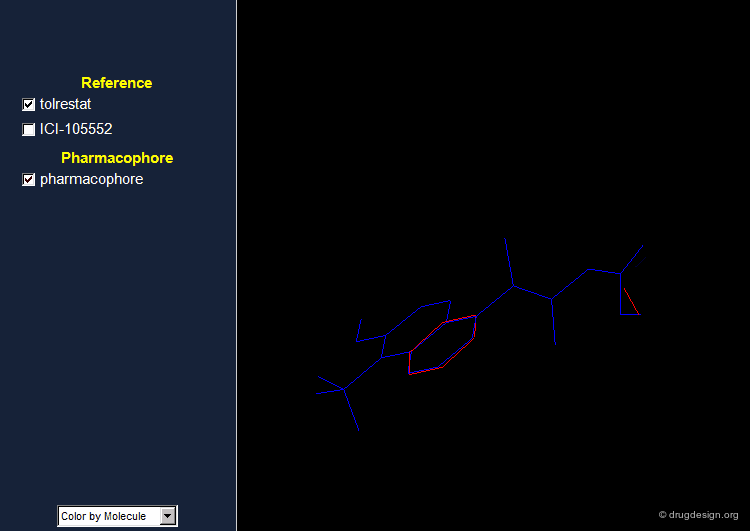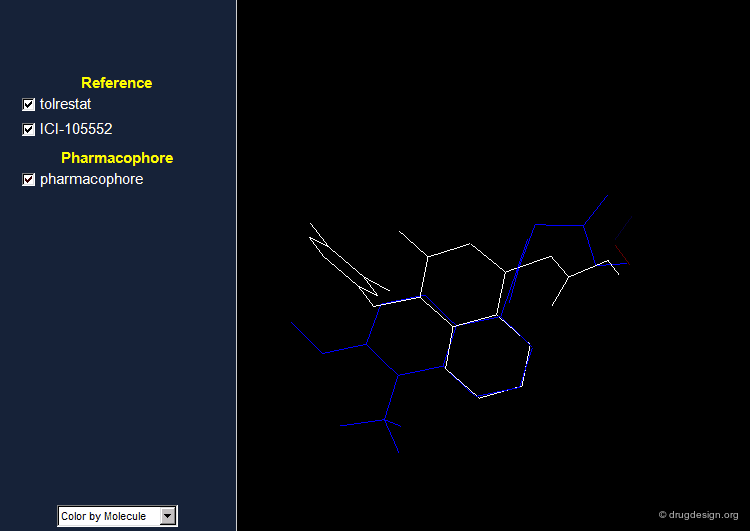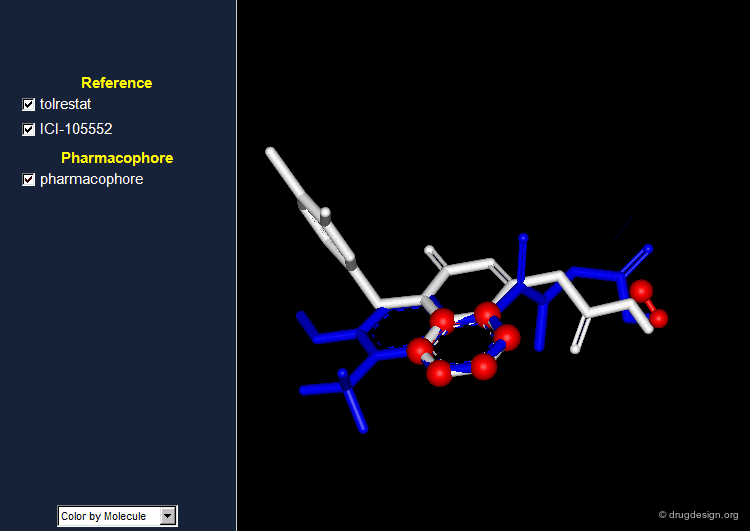
By comparing the 3D features of the two molecules, one can derive a pharmacophore model for aldose reductase inhibition. The pharmacophore consists of an acidic proton located at a distance of 5.5-5.7 Å of a phenyl ring with a height of about 1.2 Å above the plane of the aromatic ring.


Browser of Aldose Reductase Inhibitors¶
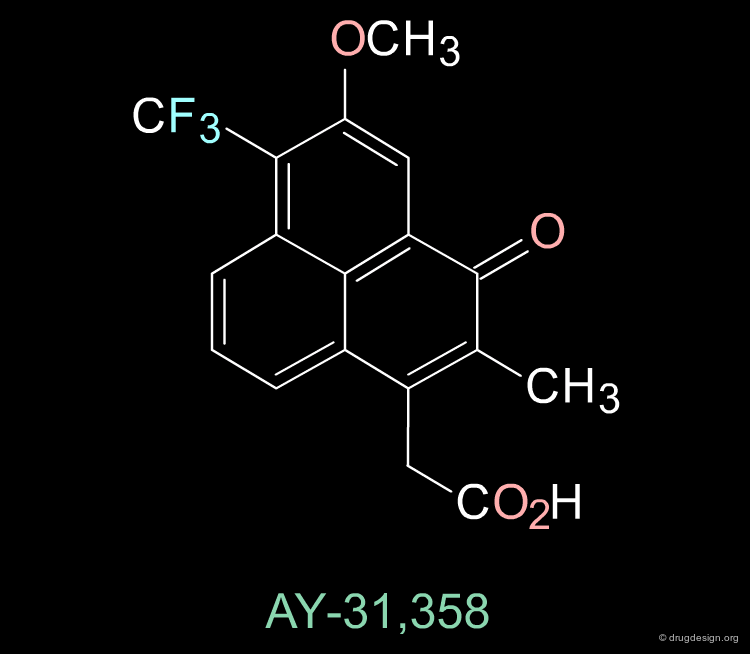
The Design of AY31358¶
The phenalene molecule AY-31,358 was designed on the basis of the pharmacophore model proposed for aldose reductase inhibitors and has shown to be a very potent inhibitor.
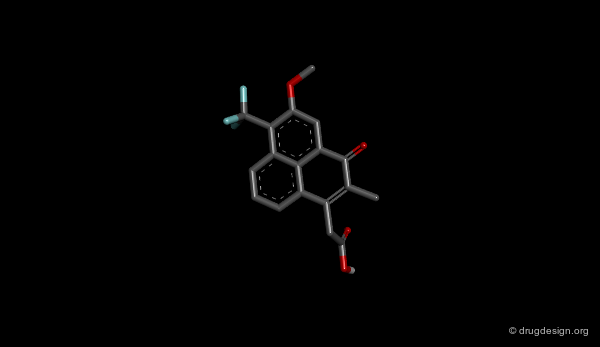

Browser of Aldose Reductase Inhibitors¶
Beta-Lactam Antibiotics¶
Biological Action of Beta-Lactam Antibiotics¶
The biological action of penicillins and cephalosporins is based on the ability of the antibiotic to irreversibly acylate the active site of the transpeptidase enzyme involved in the biosynthesis of the peptidoglycan layer of the bacterial cell wall. The bacteria is killed because the inhibition of cross-linking leads to the appearance of imperfections in its protecting 3-D wall, which eventually explodes under osmotic pressure.


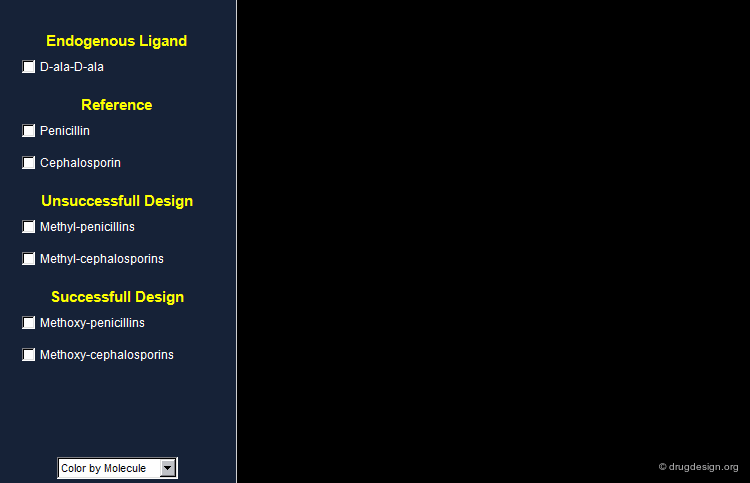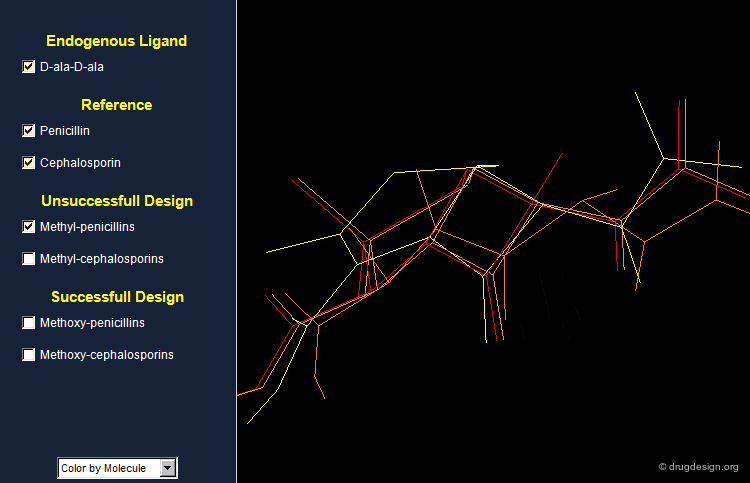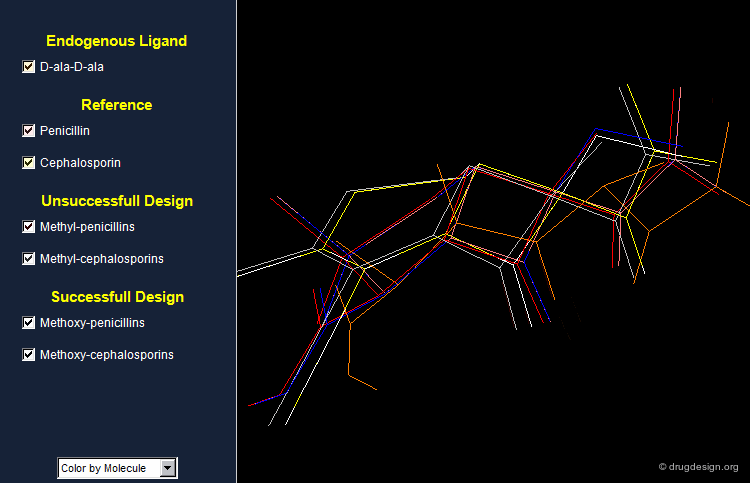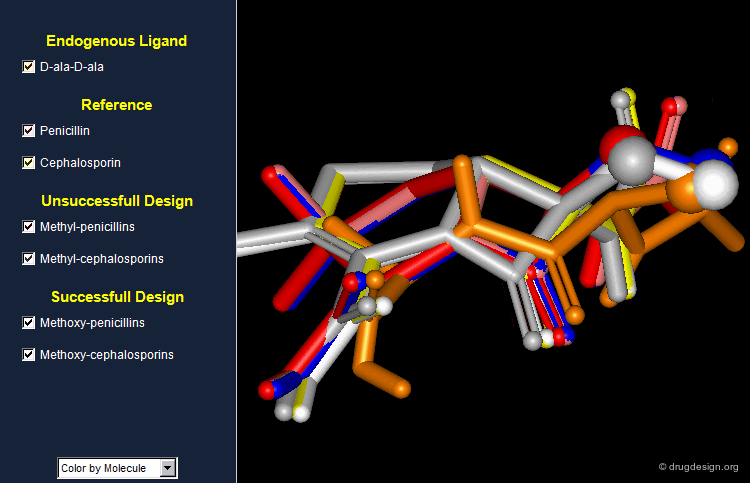
Does Penicillin Mimic an Endogenous Peptide?¶
The enzyme involved in the biosynthesis recognizes D-Ala-D-Ala terminal residues in order to carry out the cross-linking for the construction of the bacterial cell wall. This step is inhibited by the antibiotic. One can understand the antibiotic action as the drug's ability to mimic the same stereochemical features of the endogenous D-Ala-D-Ala terminal residues.

articles
Bacterial Cell Wall Synthesis and Structure in Relation to the Mechanism of Action of Penicillins and Other Antibacterial Agents Strominger JL and Tipper DJ Am. J. Med. 39 1965
Attempts to Increase Antibacterial Activities¶
Based on these considerations, it was suggested that the mimicry could be improved by introducing an additional methyl group in the penicillins to mimic the methyl residue of D-Ala-D-Ala. This was expected to reveal molecules with enhanced antibiotic activities. The methyl analogues were synthesized both in the penicillin and cephalosporin series.

articles
6-Methyl Penicillins and 7-Methyl Cephalosphorins Bohme EHW, Applegate HE, Toeplitz B, Dolfini JE and Gougoutas JZ J. Am. Chem. Soc. 93 1971
Methyl Penicillins and Methyl Cephalosporins¶
Incoporating this idea, 6a-methyl-penicillins and 7a-methyl-cephalosporins were synthesized but were practically inactive.

A Good Hypothesis with a Bad Design!¶
The methyl analogs showed poor biological activities. The design was in fact based only on 2D considerations. Looking at the structures in 3D reveals some subtle aspects that might be important. In particular, one can see that the methyl groups in the antibiotics and in D-Ala-D-Ala do not have the same orientation. This might be the reason for the weak activities observed for the methylated analogs.

An Example of a Good Hypothesis Well Exploited¶
Increasing the mimicry with D-ala-D-ala was perhaps a good idea however it was not properly exploited. Looking now at the structures in 3D indicates that a methoxy substituent (instead of a methyl) would lead to better mimics with the endogenous substrate. Indeed the methoxy analogs were synthesized both in the penicillin and cephalosporin series and showed enhanced biological activities when compared to the unsubstituted analogs.



articles
β-Lactam Antibiotics from Streptomyces Nagarajan R, Boeck LD, Gorman M, Hamill RL, Higgens CE, Hoehn MM, Stark WM and Whitney JG J. Am. Chem. Soc. 93 1971
Browser of Beta-Lactam Antibiotics¶
GABA-Uptake Inhibitors¶
Therapeutic Utility of GABA-Uptake Inhibitors¶
GABA (γ-aminobutyric acid) is a major neurotransmitter. The dysfunction of GABA-ergic synapses has been invoked in Parkinson's disease, epilepsy and schizophrenia. One of the possible ways to palliate the GABA deficiency lies in the inhibition of uptake mechanisms of this neurotransmitter.
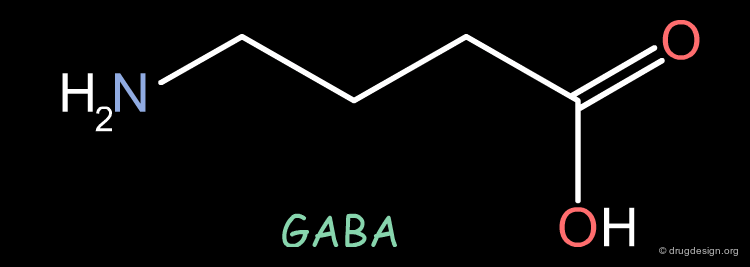

Reference Set of GABA-Uptake Inhibitors¶
Several GABA-uptake inhibitors were discovered; SKF 89976-A and SKF 100330-A represent examples of such inhibitors. The analyses of their 3D structures permitted the definition of a pharmacophore model for GABA-uptake inhibition.


Pharmacophore of GABA-Uptake Inhibitors¶
The pharmacophore derived for GABA-uptake inhibitors is the following: the amine function should be located at a distance of 4.2 Å from the center of the acidic group. The first phenyl ring is located 9.5 Å from the acidic center and 6 Å from the amino group while the second phenyl ring is at a distance of 10.5 Å from the acidic center and 6.5 Å from the amine.


Browser of GABA-Uptake Inhibitors¶

Design of a New GABA-Uptake Inhibitor¶
Based on the pharmacophore model, which was established with known GABA-uptake inhibitors, attempts were made to design new classes of inhibitors. 6-(3,3-diphenylpropyl) guvacine is a representative example and the compound showed good GABA-uptake inhibition activities.
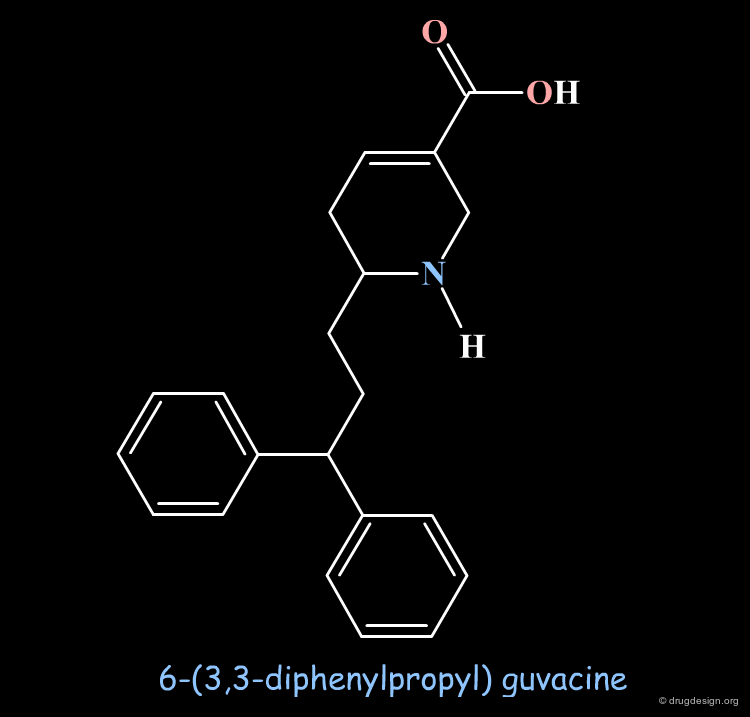

Browser of GABA-Uptake Inhibitors¶

ADDITIONAL CASE STUDIES¶
Additional Case Studies¶

Copyright © 2024 drugdesign.org