Protein Structure¶
Info
Basic principles in protein structure are illustrated in this chapter, starting with their amino acid building blocks and continuing to their folded, complex 3D architecture.
Number of Pages: 141 (±3 hours read)
Last Modified: May 2006
Prerequisites: None
Structural and Functional Diversity of Proteins¶
Proteins are Fundamental to Life¶
Proteins form the very basis of life. They are the most abundant macromolecules in living cells. Thousands of different proteins can be found in a single cell, and proteins have been identified in all parts of all cells. Virtually every property that characterizes a living organism is affected by proteins.
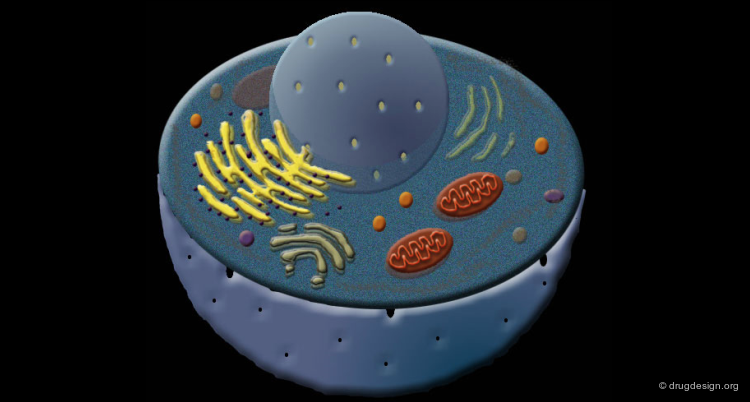
Great Diversity of Protein Biological Functions¶
The primary responsibility of proteins is to execute the tasks directed by genomic information. The proteins act like the workhorses of the cells, they display high diversity and a wide variety of specialized functions.

Chemical Nature of Proteins¶
All proteins are linear polymers made up of various combinations of 20 amino acids, and are composed mainly of C, H, O and N atoms. The amino acid sequence that defines the protein is determined by the transcription of the genetic code. Remarkably, a cell can produce proteins with different properties and activities by assembling the same 20 amino acids in many different combinations and sequences.

Structural Diversity of Proteins¶
The great diversity of the functional properties of proteins is directly related to their subtle three-dimensional structures. Every protein has a unique 3D structure that determines its function.

Link between Protein Sequence, Folding and Function¶
Importance of Protein 3D Structures¶
The formation of a protein in its biologically active form requires the folding of the protein into a precise three-dimensional structure. Changes in this structure (for example by denaturation) are consistently associated with loss of function of that protein.
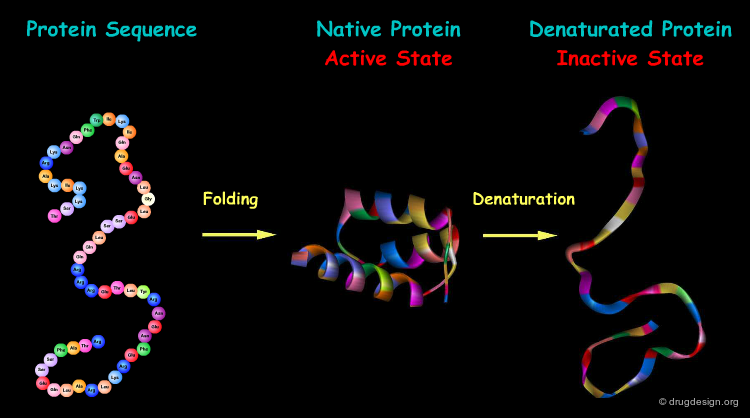
Protein Folding¶
The process by which a linear amino acid sequence of a protein is converted into a precise three-dimensional architecture is a key issue in biochemistry, and is known as the 'protein folding' problem.

book
Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding W.H. Freeman 1999
Mechanisms of Protein Folding, 2nd edition Oxford University Press 2000
Anfinsen's Dogma¶
The first and most important clue to the protein folding puzzle comes from classic experiments by Christian Anfinsen who demonstrated that denaturated ribonuclease spontaneously refolds into its active conformation in vitro. This proved that there is sufficient information in the protein sequence to determine its correct folding.

articles
Reductive Cleavage of Disulfide Bridges in Ribonuclease Sela M, White FH, and Anfinsen CB Science 125 1957
Anfinsen's Dogma and Levinthal's Paradox¶
The underlying principle that can be deduced from Anfisen's experiment is that the energetically favored state of a protein coincides with its correctly folded one. However, Levinthal demonstrated that a systematic random search of all possible conformations of the protein would take an infinite amount of time until the most stable form is found, whereas in real cells this occurs within seconds.

articles
Are There Pathways for Protein Folding? Levinthal C J. Chim. Phys 65 1968
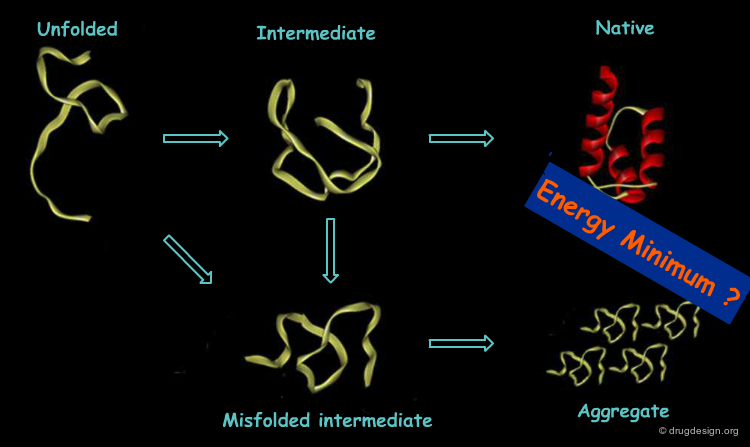
The Pathway Theory and Energy Funnels¶
The most plausible hypothesis was that folding follows a limited number of pathways, until the protein reaches the final fold. Thus, the search for a free energy minima is more like balls rolling down funnels rather than balls rolling aimlessly on a flat surface. The funnel energy landscape thus accommodates folding kinetics and thermodynamic stability.


articles
From Levinthal to Pathways to Funnels Dill KA and Chan HS Nat. Struct. Biol. 4 1997
The Fundamentals of Protein Folding: Bringing Together Theory and Experiment Dobson CM and Karplus M Curr.Opin. Struct. Biol. 9 1999
Understanding Protein Folding with Energy Landscape Theory. Part I: Basic Concepts Plotkin SS and Onuchic JN Q. Rev. Biophys. 35 2002
Navigating the Folding Routes Wolynes PG, Onuchic JN, Thirumalai D Science 267 1995
Mechanisms of Protein Folding¶
Many intricate mechanisms were put forward to describe simple pathways of protein folding. Some of these models include the initial formation of secondary structure elements, the initial formation of compact shapes by hydrophobic collapses, or even more complicated combinations of these two processes which are beyond the scope of this chapter.

articles
Is Protein Folding Hierarchic? I. Local Structure and Peptide Folding Baldwin RL and Rose GD Trends Biochem. Sci. 24 1999
Is Protein Folding Hierarchic? II. Folding Intermediates and Transition States Baldwin RL and Rose GD Trends Biochem. Sci. 24 1999
Protein Folding Mechanisms: New Methods and Emerging Ideas Brockwell DJ, Smith DA, and Radford SE Curr. Opin. Struct. Biol. 10 2000
Topological and Energetic Factors: What Determines the Structural Details of the Transition State Ensemble and {quote}En-Route{quote} Intermediates for Protein Folding? An Investigation for Small Globular Proteins Clementi C, Nymeyer H, and Onuchic JN J. Mol. Biol. 298 2000
From Levinthal to Pathways to Funnels Dill KA and Chan HS Nat. Struct. Biol. 4 1997
Polymer Principles and Protein Folding Dill K.A Protein Sci. 8 1999
Is there a Unifying Mechanism for Protein Folding? Daggett V and Fersht AR Trends Biochem. Sci. 28 2003
The Fundamentals of Protein Folding: Bringing Together Theory and Experiment Dobson CM and Karplus M Curr.Opin. Struct. Biol. 9 1999
Protein Folding and Unfolding at Atomic Resolution. Fersht AR and Daggett V Cell 108 2002
Protein Folding: from the Levinthal Paradox to Structure Prediction Honig B J. Mol. Biol. 293 1999
Understanding Protein Folding with Energy Landscape Theory. Part I: Basic Concepts Plotkin SS and Onuchic JN Q. Rev. Biophys. 35 2002
Navigating the Folding Routes Wolynes PG, Onuchic JN, Thirumalai D Science 267 1995
The Protein Misfolding Problem¶
In real cells the protein folding process seems to be more complex. An unexpected mechanism was observed where proteins called "chaperons" were discovered, whose role is to keep proteins from getting out of their right folding path. This finding cast doubt on the validity of Anfinsen's thermodynamic model under cellular conditions and prompted the search of improved models.
articles
The Unfolding Puzzle of Protein Folding King J Tech. Rev. 96 1993
Misfolding the Way to Disease Taubes G Science 271 1996
Defective Protein Folding as a Basis of Human Disease Thomas PJ, Qu BH, and Pedersen PL Trends Biochem. Sci. 20 1995
Molecular Chaperones - Cellular Machines for Protein Folding Walter S and Buchner J Angew. Chem. Int. Ed Engl. 41 2002
Challenge in Understanding Protein Structure¶
Despite the exciting advances in our understanding of protein folding in the last decade, it remains an unresolved enigma. The prediction of the 3D structure of a protein from sequence data alone is a formidable challenge.

articles
Protein Structure Prediction and Structural Genomics Baker D and Sali A Science 2001 294
Protein Folding: from the Levinthal Paradox to Structure Prediction Honig B J. Mol. Biol. 293 1999
Amino Acids: Building Blocks of Proteins¶
Amino acids: Building Blocks of Proteins¶
Amino Acids are the building blocks of proteins. All proteins, from the most ancient lines of bacteria to the most complex forms of life, are constructed with the same basic set of 20 amino acids existing in nature. In the following view you can see the full name of each amino acid as well as its three-letter and one-letter codes.

α-Amino Acids¶
The amino acids found in proteins have the following general topology called the α-structure. An α-amino acid is characterized by a central carbon atom (called the Cα), which is connected to the following four groups: (1) amino; (2) carboxyl; (3) hydrogen atom and (4) distinctive R group - often referred to as the "side chain".

α-Amino Acid Stereoisomers¶
With four different groups connected to the central sp3 carbon α, α-amino acids are chiral and possess two stereoisomers: the L and the D enantiomers. The amino acids in nearly all natural proteins have the L stereochemistry.

articles
D-Amino acid Residues in Peptides and Proteins Mitchell JB and Smith J Proteins 50 2003
Diversity of the Properties of Amino Acids¶
Each of the twenty amino acids has unique physico-chemical properties depending on the nature of its side chain (R group). The amino acid side chains vary in size, shape, charge, hydrogen bonding capacity, hydrophobic character and chemical reactivity.

Amino Acids Properties¶
The following browser provides the 2D and 3D structure of all 20 standard amino acids, together with their main properties.

Classification of Amino Acids Properties¶
The amino acids can be classified and organized in different groups, based on their various physico-chemical properties as indicated in the following diagram.

Non-Standard Amino Acids¶
In addition to the standard set of twenty amino acids that are found in all proteins, other amino acids have been found in certain types of proteins. Some examples are shown here. Most of them are the result of biochemical transformations of a standard amino acid, after its incorporation into the protein sequence.

articles
How Selenium has Altered Our Understanding of the Genetic Code Hatfield DL and Gladyshev VN Mol. Cell. Biol. 22 2002
Biochemistry of Hydroxyprolines Kutton R and Radhakrishnan AN Adv. Enzymol. 37 1973
The Many Faces of Histone Lysine Methylation Lachner M, Jenuwein T Curr. Opin. Cell. Biol. 14 2002
Gamma-Carboxyglutamate Containing Proteins and the Vitamin K Dependent Carboxylase Vermeer C Biochem. J. 266 1990
From Amino Acids to Proteins¶
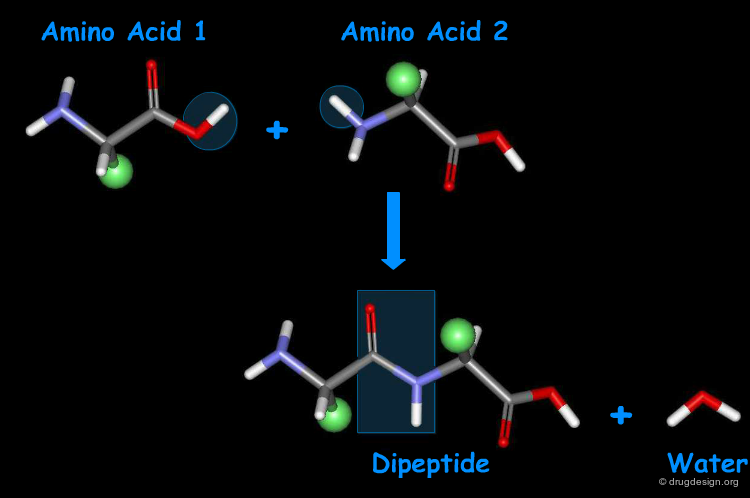
Amino Acids are Linked by Peptide Bonds¶
Amino acids can be covalently linked together by the formation of an amide bond between the α-amino group of one amino acid and the α-carboxyl group of another amino acid. The resulting amide bond is known as "peptide bond". The formation of a dipeptide from two amino acids in a condensation reaction is accompanied by the removal of a water molecule.
Peptide Biosynthesis¶
In the cell, amino acids are chemically linked by the biochemical process of translation, or protein biosynthesis. The polymer biosynthesis is a template-directed reaction catalyzed by the ribosome.
book
Lehninger AL, Nelson DL and Cox MM Principle of Biochemistry, 2nd edition Worth
Polymer Amino-Acids¶
A series of amino acids joined by peptide bonds form a polypeptide chain. Each amino acid unit is called a "residue". A polypeptide chain consists of a regularly repeating part, called the "main chain" or the "backbone" and a variable part, comprising the distinctive side chains.
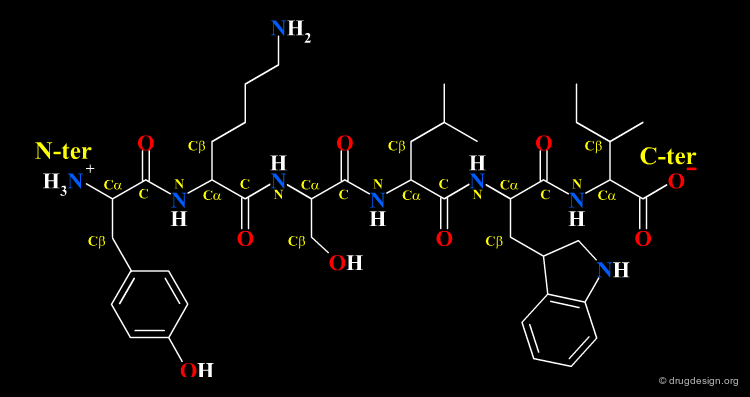




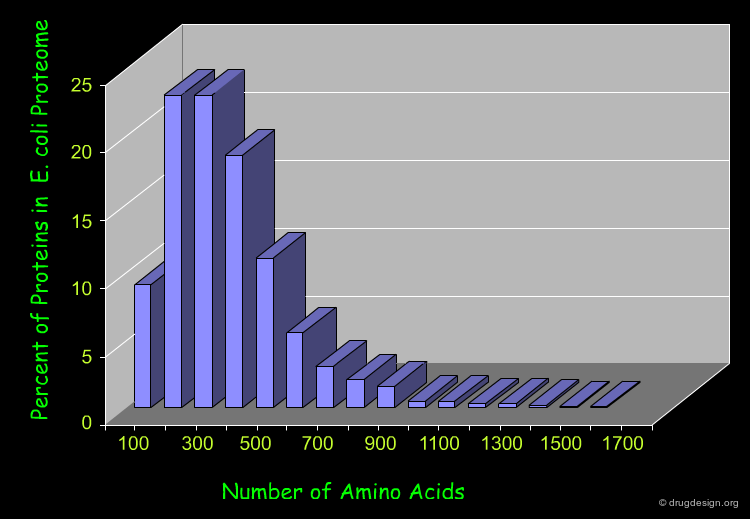
Length of Proteins¶
Most natural polypeptide chains contain between 50 and 2000 amino acid residues. Shorter chains are called oligopeptides or simply peptides. The following histogram shows the length of the proteins in the E-coli proteome.
More than One Polypeptide Chain¶
Some proteins contain a single polypeptide chain, whereas others may aggregate to form dimeric, trimeric and multimeric clusters. The polypeptide chains in these clusters can be either identical or different.

Conjugated Proteins¶
There are proteins that incorporate non-peptidic molecules in their overall structure, either bonded covalently or positioned by other forces. These are called conjugated proteins, and the non-peptide components are referred to as prosthetic groups. The prosthetic moieties play an essential role in protein function. Conjugated proteins are classified according to the chemical nature of their prosthetic group. For example:

Examples of Conjugated Proteins¶
Examples of known structures of conjugated proteins are shown here. The protein is visualized by a ribbon representation with relevant residues represented by sticks. The prosthetic group is shown in a "ball and stick" representation.

Pdb
Glycoprotein, PDB entry: 1IVO
Nucleoprotein, PDB entry: 1CWP
Flavoprotein, PDB entry: 2FX2
Metalloprotein, PDB entry: 3CPA
Phosphoprotein, PDB entry: 3JST
Hemoprotein, PDB entry: 2HHB
Cross-Linked Polypeptide Chains¶
In some proteins, two cysteine side chains are located close together so that their thiol groups can be oxidized to produce a covalently dimeric amino acid joined by a disulfide bond (bridge). This dimeric amino acid is known as "Cystine". The covalent cross-linking helps to stabilize the protein structure. Intramolecular disulfide bonds are mainly found in proteins that are in the extracellular space (e.g. digestive enzymes, hormones).

articles
Disulfide Bond Formation, a Race between FAD and Oxygen Bardwell JC Dev. Cell. 3 2002
Thiol/disulfide Exchange Equilibria and Disulfide Bond Stability Gilbert HF Methods Enzymol. 251 1995
Mechanisms and Catalysts of Disulfide Bond Formation in Proteins Creighton TE, Zapun A and Darby NJ Trends Biotechnol. 13 1995
Native Disulfide Bond Formation in Proteins Woycechowsky KJ and Raines RT Curr. Opin. Chem. Biol. 4 2000
Geometry of Proteins and Peptides¶
Peptide Bonds are Planar¶
The peptide bond is essentially planar. All the atoms in the central amide bond lie in the same plane.

Why the Peptide Bond is Planar?¶
The chemical nature of the peptide bond explains this geometric preference. The lone pair on the nitrogen atom can delocalize into the adjacent carbonyl as illustrated in the following resonance structure. Thus, the peptide bond has a considerable double-bond character, which prevents free rotation about this bond and favors a planar geometry.

Cis and Trans Isomers of the Peptide Bond¶
The restricted rotation about the planar peptide bond allows the amide moieties to exist as discrete "s-cis" and "s-trans" isomers. In proteins, the great majority of peptide bonds are s-trans.
![]()
articles
Non-Proline Cis Peptide Bonds in Proteins Jabs A, Weiss MS and Hilgenfeld R J. Mol. Biol. 286 1999
Trans Isomer Favored¶
The preference of the s-trans over the s-cis isomer can be explained by steric clashes between the amino acids side chains attached to the α-carbon which hinder formation of the s-cis isomer. In other words, the s-trans isomer is intrinsically favored energetically due to fewer repulsions between non-bonded atoms.

Isomers of Proline¶
For proline the situation is different because the steric differences between cis and trans isomers are minimal. In folded proteins about 10% of the proline-peptide bonds are found in the cis conformation.

Peptide Torsion Angles¶
The torsion angles along the polypeptide chain are called omega (Ω) (the peptide bond), phi (Φ) and psi (Ψ) (see view below). Unlike the peptide bond, the bonds defined by the torsion angles Φ and Ψ are single Σ-bonds and can rotate.
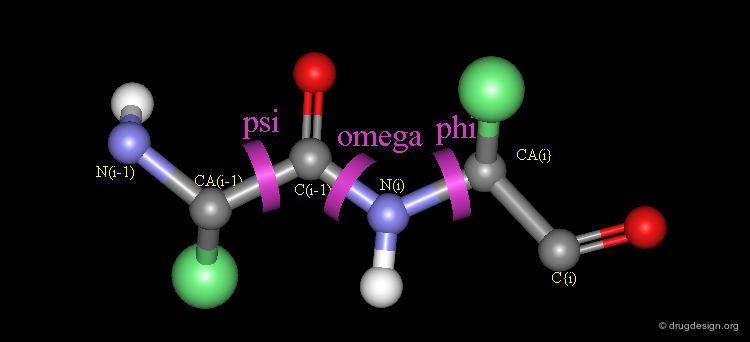
Conformational Freedom¶
The free rotation about two rotatable bonds of each amino acid allows adjacent rigid peptide units to rotate about these bonds, and as a consequence the polypeptide chain can fold in many different ways. The exact geometry of a polypeptide chain is defined by the respective Φ and Ψ values.

Conformational Complexity of Polypeptide Chains¶
With two rotatable bonds for each amino acid, the conformational complexity of any polypeptide chain is huge. Just taking the three energetically favored staggered conformation for each rotatable bond yields the following results:

Not All Φ/Ψ Torsion Angles are Possible¶
Whereas there is considerable conformational freedom around the Φ and Ψ angles due to steric repulsions, not all torsion angles are possible. For example, in the situation illustrated below the combination of Ψ=180 and Φ=0 is "disallowed" due to steric clashes between the two carbonyl groups.

The Ramachandran Plot¶
G.N. Ramachandran used computer models of small polypeptides to systematically explore possible Φ and Ψ combinations in polypeptide chains. Each combination of the two torsion angles was structurally examined to identify favorable interactions or steric clashes between atoms. The distribution of Φ and Ψ angles can be plotted in two-dimensions in the so-called Ramachandran plot (see next pages).

articles
Microfolding: Conformational Probability Map for the Alanine Dipeptide in Water from Molecular Dynamics Simulations Anderson AG and Hermans J Proteins 3 1988
Phi/Psi-Chology: Ramachandran Revisited Kleywegt GJ and Jones TA Structure 4 1996
On Residues in the Disallowed Region of the Ramachandran Map Pal D and Chakrabarti P Biopolymers 63 2002
Conformation of Polypeptides and Proteins Ramachandran GN, Sasisekharan V Adv. Protein. Chem. 23 1968
Φ and Ψ Distribution¶
For all amino acids except glycine, the following Ramachandran plot describes the allowed and disallowed Φ-Ψ torsion angle combinations. The white areas correspond to disallowed conformations (where atoms clash). The blue areas correspond to allowed conformations. Borderline regions in which the atoms are allowed to come a little bit closer together appear in light blue.

Interactive Ramachandran Plot¶
The following is an interactive Ramachandran plot for Ala-Ala-Ala tripeptide. Each point on the plot shows the Φ and Ψ values for the central Ala residue in the tripeptide. By clicking on a given point in the diagram you can visualize the corresponding 3D structure.
Torsion Angles Observed in Proteins¶
By displaying all sets of Φ/Ψ torsion angles observed in a given protein (here, the lysozyme protein) on top of a Ramachandran plot, the fit between the predicted and observed values can be checked (note that glycine residues are not included in this diagram).

Glycine Residue Torsion Angles¶
In the case of glycine the situation is different because this residue has no side chain, which allows for a broader range of Φ/Ψ values. Including the glycine residues torsion angles of the protein considered (lysozyme) on top of the general Ramachandran plot (pink triangles), often results in Φ/Ψ values that are located in the seemingly disallowed area.

Side Chain Conformations¶
For similar reasons (as for the geometry of the backbone) the conformational possibilities of the side chains are also restricted. The distinct, low energy conformations of a given residue are called side chain rotamers. For example the nine rotamers of the side chain of aspartate are shown here.

articles
Conformation of Amino Acid Side-Chains in Proteins Janin J and Wodak S J. Mol. Biol. 125 1978
Tertiary Templates for Proteins. Use of Packing Criteria in the Enumeration of Allowed Sequences for Different Structural Classes Ponder JW and Richards FM J. Mol. Biol. 193 1987
Side Chain Atomic and 3D Nomenclature¶
The side chain atoms of amino acids and the corresponding torsion angles are called by letters of the Greek alphabet. Here, for example, the Lysine residue is shown. Amino acids may have up to five possible torsions within their side chain, depending on the structure of the residue.
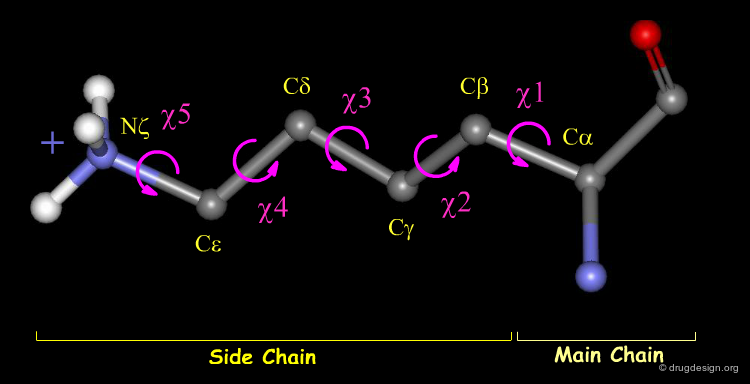
Side Chain Conformations¶
In principle, the torsion angles adopted by side chain residues should be close to theoretical equilibrium values, which minimize close contacts between adjacent atoms of the residue (e.g. staggered in the case of a sp3-sp3 bond and eclipsed for a sp2-sp3 bond). However, some combinations of torsion angles can create unfavorable interactions between distant atoms. For example a staggered conformation of the leucine residue with steric clashes is shown in the view below (for clarity hydrogen atoms have been omitted).
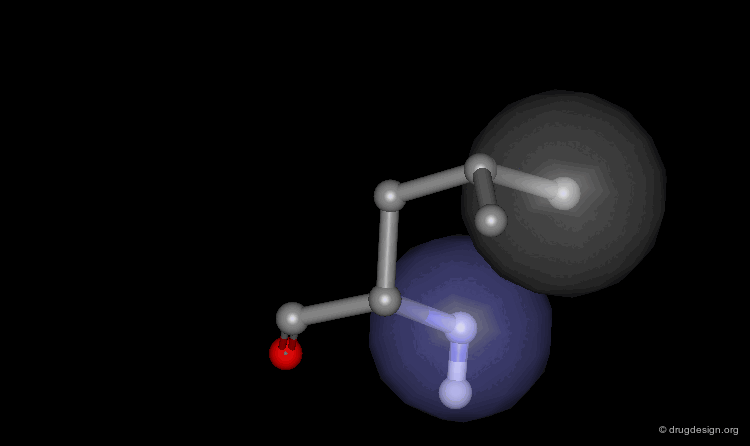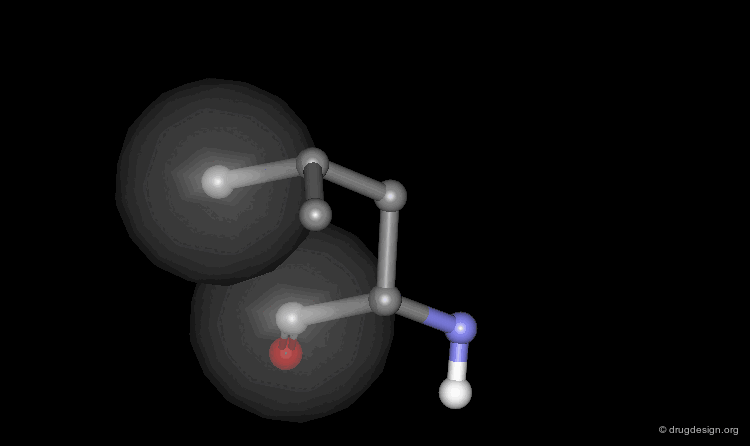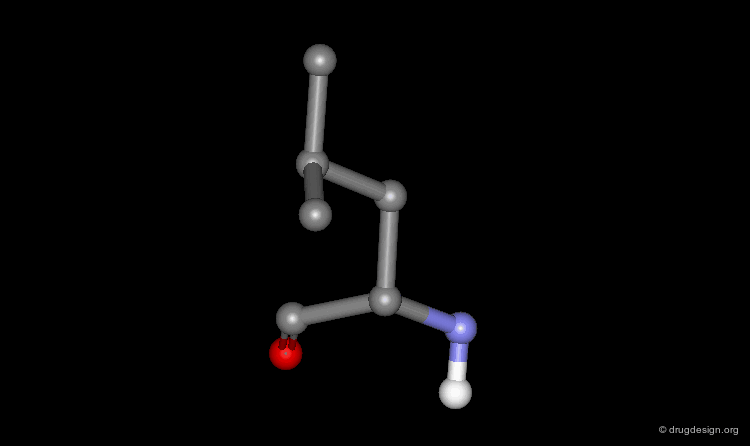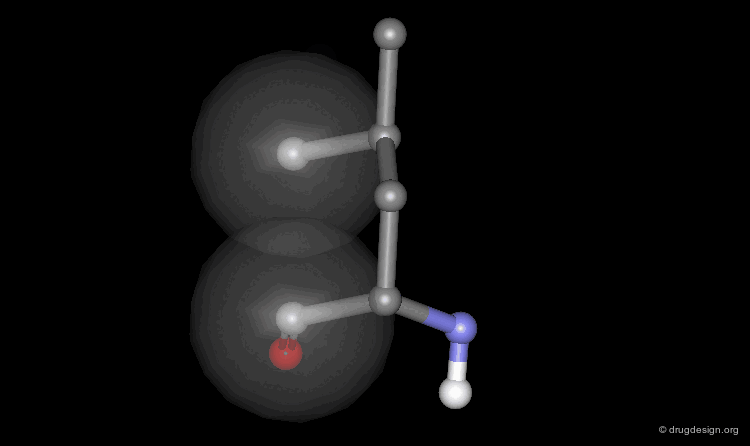
Non-Rotameric Side Chain Conformations¶
The side chain conformations are highly affected by external parameters, such as ligand binding and protein packing. Consequently, commonly up to 10% of the side chains in a protein adopt non-regular (rotameric) conformations. For example, in the following enzyme non-rotameric side chains are shown with green carbon atoms. Note that three of these residues were found to interact with the ligand molecule (white) in the complex.

articles
Strain in Protein Structures as Viewed through Nonrotameric Side Chains: I.Their Position and Interaction. Heringa J and Argos P Proteins 37 1999
Strain in Protein Structures as Viewed through Nonrotameric Side Chains: II. Effects Upon Ligand Binding Heringa J and Argos P Proteins 37 1999
Rotamers: to be or not to be? An Analysis of Amino Acid Side-Chain Conformations in Globular Proteins Schrauber H, Eisenhaber F, and Argos P J. Mol. Biol. 230 1993
Protein Structure Overview¶
Protein Structure Complexity¶
The most striking feature of the folded conformation of a protein is its complexity, revealing the organization and the relationships of thousands of atoms in three-dimensional space. We will ease our way into the confusing jumble of atomic coordinates, distances, torsions angles and interactions with a short overview of protein architecture.

The Four Levels of Protein Architecture¶
There are four levels in the hierarchy of the protein structure that represent different levels of structural complexity. They correspond to the primary, secondary, tertiary and quaternary levels of the protein structure.
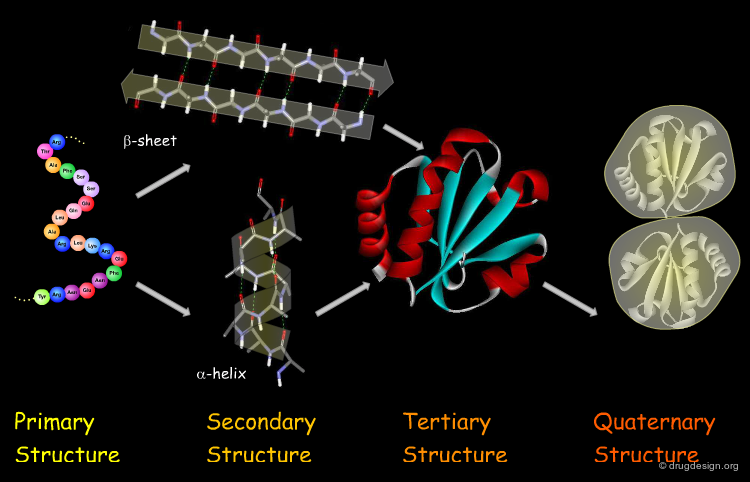
Primary Structure¶
The first and most fundamental level of a protein is its primary structure, which describes the atomic connectivities of the protein. The primary structure is defined by the amino acid sequence and may include cystines that are formed during cross-linking.
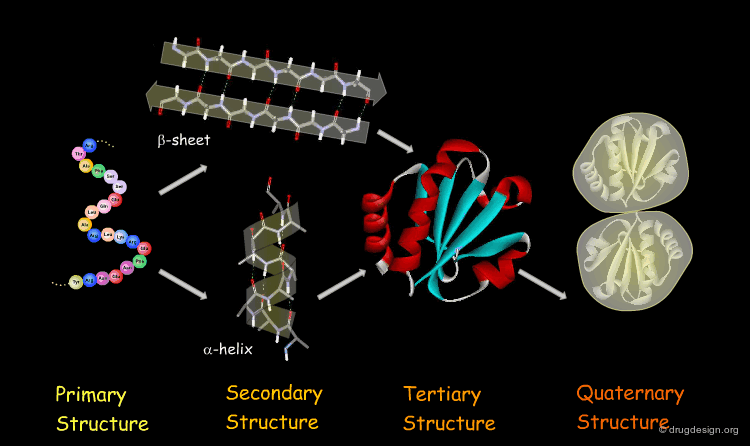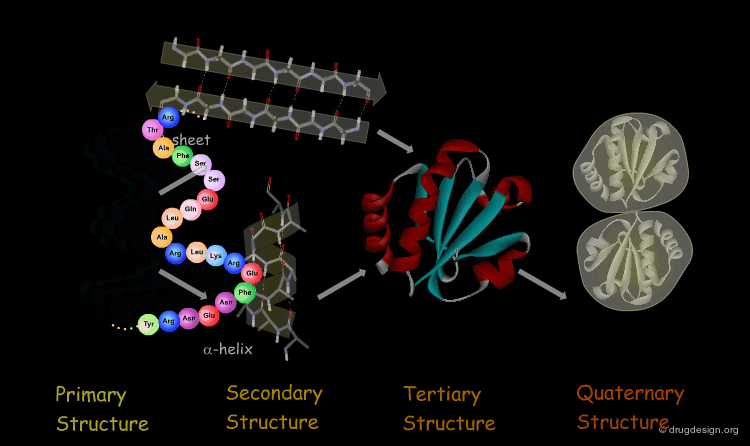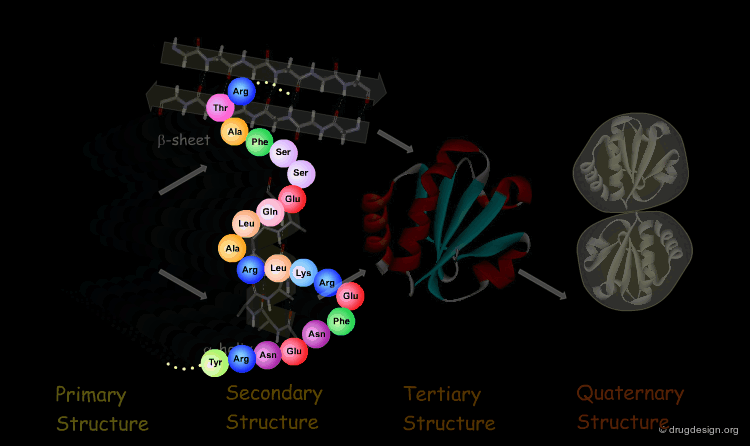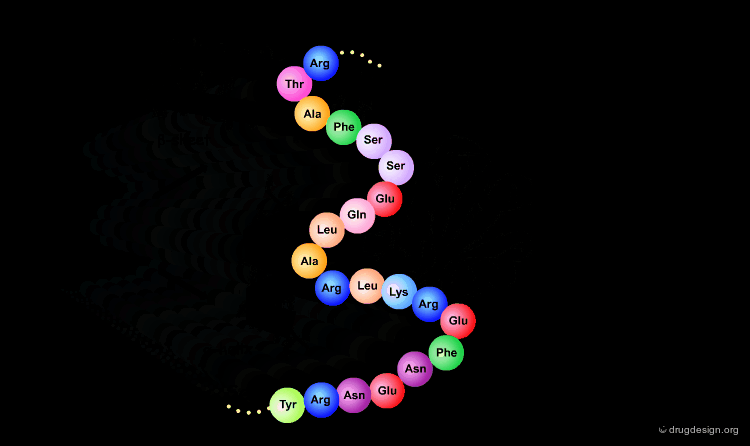
Secondary Structure¶
The next level of protein structure, the secondary structure, includes local regular conformations of the polypeptide chain. The two major elements of the secondary structure are the α-helix and the β-sheet.

Tertiary Structure¶
The tertiary structure of a protein is the overall 3D architecture of the folded polypeptide chain which also includes the assembly of the various secondary structure elements in 3D.

Quaternary Structure¶
Finally, proteins consisting of more than one polypeptide chain display a quaternary structure, which refers to the spatial relationship between the individual polypeptide chains (subunits).

Forces Involved in Protein Stability¶
Protein stability arises from many noncovalent interactions such as hydrogen-bonding, hydrophobic and electrostatic interactions. Their individual forces are weak (many hundred times weaker than the strength of a covalent bond), but their great number and broad distribution create a cooperative effect that dictates how the protein fold into a conformation.

Proteins are not Static¶
Proteins are not motionless under physiological conditions. Thermodynamic vibrations of the atoms may induce movement of entire regions of the protein. Dynamic changes of protein conformations range from local side chain rearrangements to complex backbone motions such as the hinge bending illustrated below. Conformational changes are key elements for the biological function of the protein.

articles
MolMovDB: Analysis and Visualization of Conformational Change and Structural Flexibility Echols N, Milburn D, and Gerstein M Nucleic Acids Res. 31 2003
The Morph Server: a Standardized System for Analyzing and Visualizing Macromolecular Motions in a Database Framework Krebs WG and Gerstein M Nucleic Acids Res. 28 2000
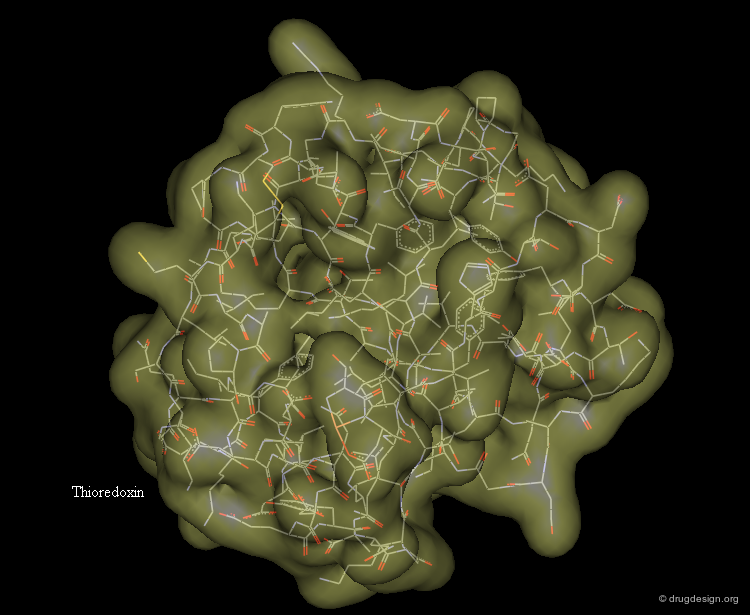
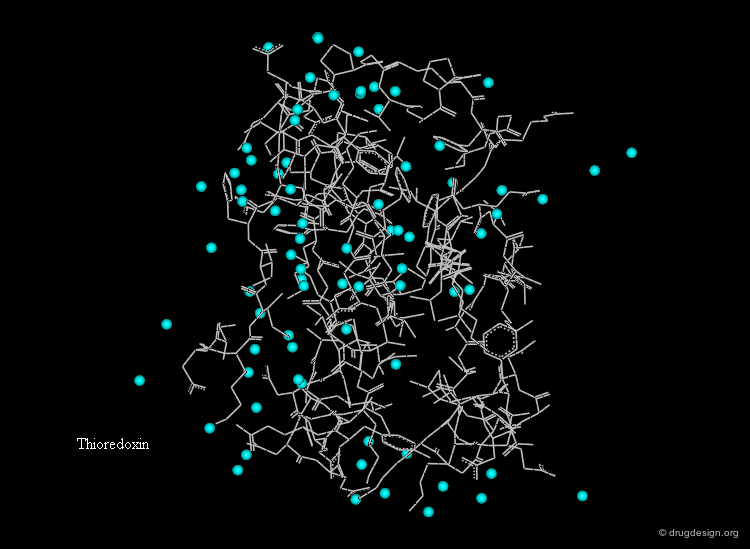
Representing Protein Structures¶
Various representations are used to visualize protein structures, each type highlighting a specific feature of the considered structure. The most common visualization techniques are illustrated here using the thioredoxin protein as an example.

Wireframe Representation¶
The wireframe representation consists of a visualization of the bonding arrangement of the atoms of the protein in 3D (for clarity non-essential hydrogen atoms are not included). In all visualization softwares an additional functionality displays the hydrogen bonds. The coloring scheme is the following: carbon (grey), oxygen (red), nitrogen (blue) and sulfur (yellow).

Ball and Stick Representation¶
Interesting details of protein folding can be revealed by removing the side chains and only displaying the heavy atoms of the backbone. Here, the backbone is displayed as an assembly of atoms and the bonds are visualized in a "ball and stick" representation.

Cα Trace Representation¶
A simpler way to visualize the general fold of a polypeptide chain is to use a Cα trace. The Cα trace representation is a line that connects the α carbon positions of each amino acid residue (note that the "bonds" shown here do not correspond to real ones).

Ribbon Representation¶
The ribbon representation provides an overview of the overall folding of the protein. α-Helices and β-sheets are easily recognized. The coloring scheme here shows the various secondary structures.

Cartoon Representation¶
This is a cartoon-type representation showing the folding of the protein in a Cα trace framework. In this type of visualization α-helices are represented as cylinders and β-strands as flat arrows.
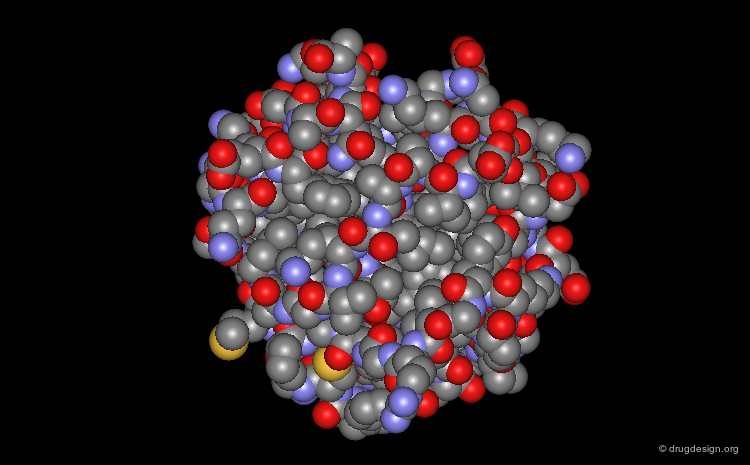
Space Filling - CPK Representation¶
The atoms are shown here as CPK spheres with their volume corresponding to the van der Waals radii of the atom.
Surface Representation¶
The surface representation visualizes the molecular envelope of the protein. Moreover, a color-code property can be used for the graphical representation of the surface (in the example below: the electrostatic potential).

Primary Structure¶
Primary Structure¶
The primary structure of a protein accounts for its covalent structure. Thus, the primary structure is a linear description of the sequence of amino acids of the protein (and may include post-translational modifications, such as glycosylation, phosphorylation and the locations of disulfide bonds). The relative spatial arrangement of the linked amino acids is not specified in this context.

Unique Primary Structure for Each Protein¶
In 1953 Frederick Sanger determined the first amino acid sequence of a protein (it was for the hormone insulin). He showed that proteins have a unique amino acid sequence. All molecules of a given protein are identical, and the sequence of a protein is unique.

Primary Sequence and Protein Properties¶
The amino acid sequence of a protein is genetically determined. It governs all its chemical and biological properties and can be considered as carrying indirect information on higher levels of the protein architecture. Moreover, it can be seen as a link between the genetic message in the DNA and the three-dimensional structure that preforms the biological function of the protein.

Secondary Structure¶
Secondary Structure¶
The secondary structure refers to recurring local conformations of the polypeptide chain backbone. The two major elements of the secondary structure are the α-helix and the β-sheet.

Periodic and Non Periodic Secondary Structure Elements¶
The concept of regular repeating elements was initially introduced by Linus Pauling and Robert Corey when they proposed periodic elements for the α-helix and for the β-sheet. Subsequently, other local non-periodic structural elements, such as turns and specific loops were identified.

articles
Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain Pauling L, Corey RB, and Branson HR Proc. Natl. Acad. Sci.USA 37 1951
Configurations of Polypeptide Chains with Favored Orientations around Single Bonds: Two New Pleated Sheets Pauling L, Corey RB Proc. Nat. Acad. Sci. USA 37 1951
Hydrogen Bonds in Secondary Structure Elements¶
All the secondary structure elements are folded by linking the C=O and N-H groups of the backbone together by means of hydrogen bonds. Experimental secondary structures are usually slightly distorted when compared to their ideal theoretical geometry, however the hydrogen bond pattern is still maintained.

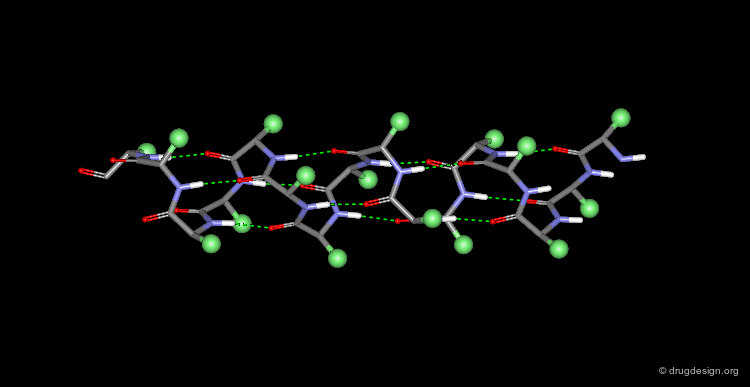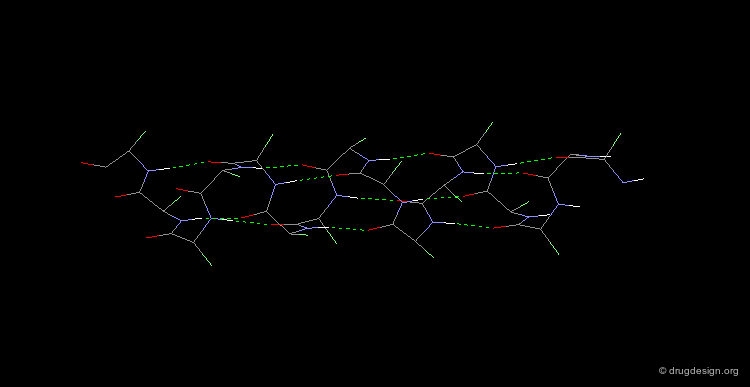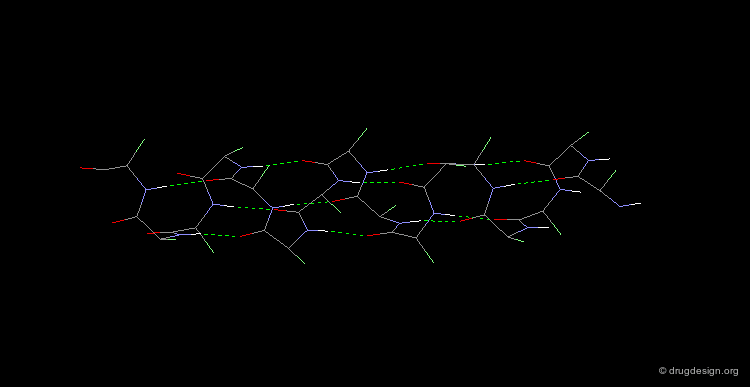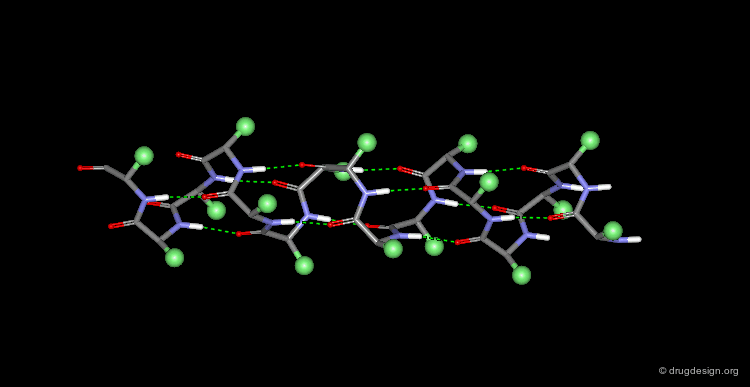
The α-Helix¶
The α-helix is the best known and most abundant form of local regular structures found in proteins. In this structure the C=O and N-H groups of all the peptide units are hydrogen-bonded. The C=O hydrogen bond acceptors and their corresponding N-H donor groups are separated by four amino acids (i.e. Oi and Ni+4) in the polypeptide chain.
articles
Helix Geometry in Proteins Barlow DJ and Thornton JM J. Mol. Biol. 201 1988
Flexibility of alpha-Helices: Results of a Statistical Analysis of Database Protein Structures Emberly EG, Mukhopadhyay R, Wingreen NS and Tang C J Mol Biol. 327 2003
General Architecture of the alpha-Helical Globular Proteins Murzin AG and Finkelstein AV J. Mol. Biol. 204 1988
Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain Pauling L, Corey RB, and Branson HR Proc. Natl. Acad. Sci. USA 37 1951
Packing of alpha-Helices: Geometrical Constraints and Contact Areas Richmond TJ and Richards FM J. Mol. Biol. 119 1978
Packing of the α-Helix¶
By rotating the α-helix visualized here you can see the tight pack of backbone atoms. The side chains (represented by green spheres in the Cβ position) extend outwards, also in a helical array.

Φ and Ψ Torsion Angles of the α-Helix¶
The α-helix can be defined by its Φ and Ψ torsion angles. When all of the residues of a polypeptide stretch have Φ and Ψ angles of approximately -57° and -47° respectively, these residues form an α-helix.


Two Enantiomeric α-Helices¶
In fact two enantiomeric α-helix structures can be defined: the right-handed (clockwise) and left-handed (counterclockwise) helices (with Φ/Ψ values of -57°/-47° and +57°/+47°, respectively). The right-handed α helix is by far the most abundant form found in proteins.


Geometry Described with Pitch and Rise¶
The helical structure repeats itself every 360°, in which the distance translated along the helical axis is 5.4 Å, which is the "pitch" of the helix. α-Helices have 3.6 amino acid residues per turn. That means that the distance translated along the helical axis for one residue is 5.4/3.6 or 1.5 Å: thus the α-helix has a rise per residue of 1.5 Å (see figure below).

Helix Macro-Dipole¶
In an α-helix the peptide planes are roughly parallel to the helix axis; the dipoles of the peptide bonds are therefore aligned in the same direction. The cumulative effect results in a substantial macro-dipole for the helix. The charges associated to the macro-dipole are about 0.5-0.7 electronic units, with a positive value at the amino end, and a negative charge at the carboxyl end.

articles
The alpha-Helix Dipole and the Properties of Proteins Hol WGJ, van Duijnen, PT and Berendsen, HJC Nature 273 1978
The Role of the alpha-Helix Dipole in Protein Function and Structure Hol WGJ Prog. Biophys. Mol. Biol. 5 1985
The alpha-Helix as an Electric Macro-Dipole Wada A Adv Biophys
1976
Amphipathic Character of the α Helix¶
Many α-helices are amphipathic, i.e. they consist of hydrophobic non-polar side chains along one side of the helical cylinder, and hydrophilic polar residues along the other side. These helices have a hydrophobic moment and they often aggregate with other hydrophobic surfaces. The amphipathic nature of a helix is best seen by use of a helical wheel, which is a projection of the helix looking down at the main axis.
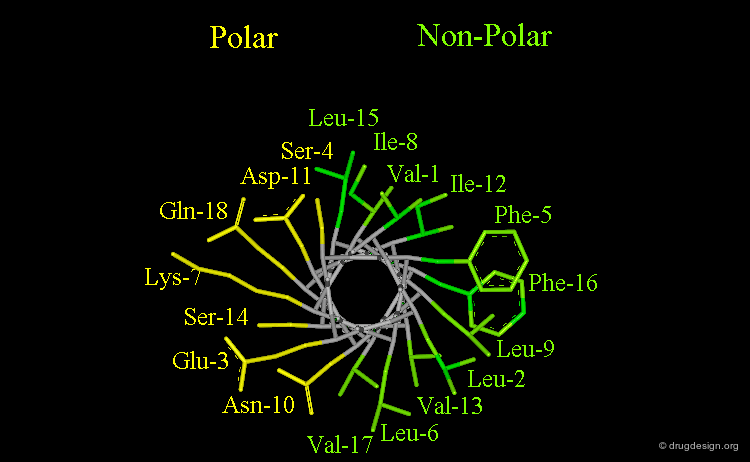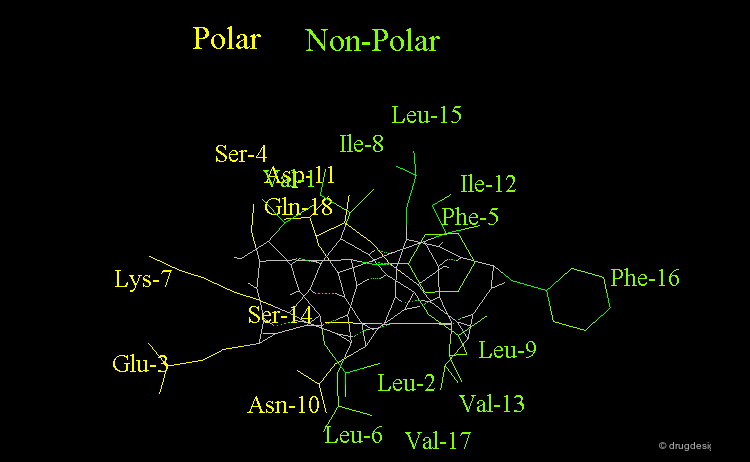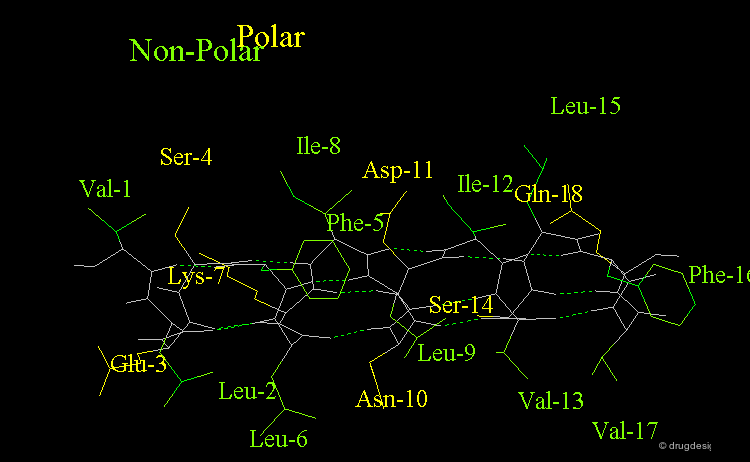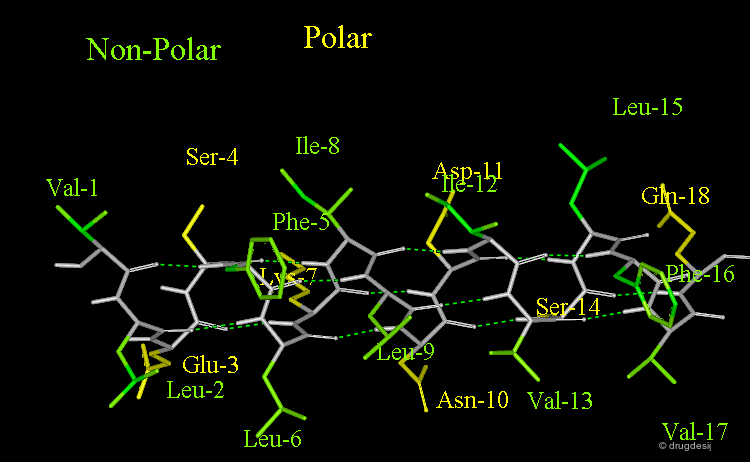
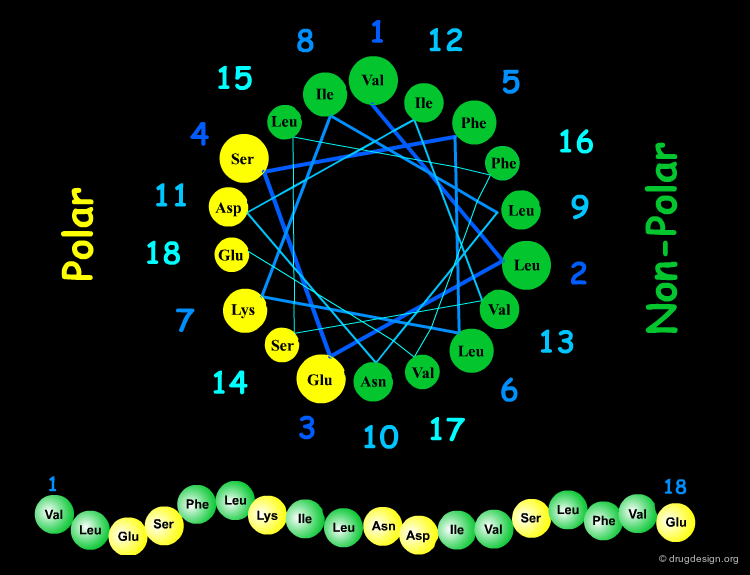
articles
Hydrophobicity Scales and Computational Techniques for Detecting Amphipathic Structures in Proteins Cornette, JL, Cease KB, Margalit H, Spouge JL, Berzofsky JA and DeLisi C J. Mol. Biol. 195 1987
The Helical Hydrophobic Moment: a Measure of the Amphiphilicity of a Helix Eisenberg D, Weiss RM, and Terwilliger TC Nature 299 1982
3(10)-Helix and π-Helix¶
There are at least three well-identified regular helices types in proteins. All the helices visualized here consist of a string of 16 amino acids colored according to the atom types with the side chains represented as green spheres. The following view shows the ideal right-handed α-helix, the more stretched 310 helix, and the more compact π-helix.
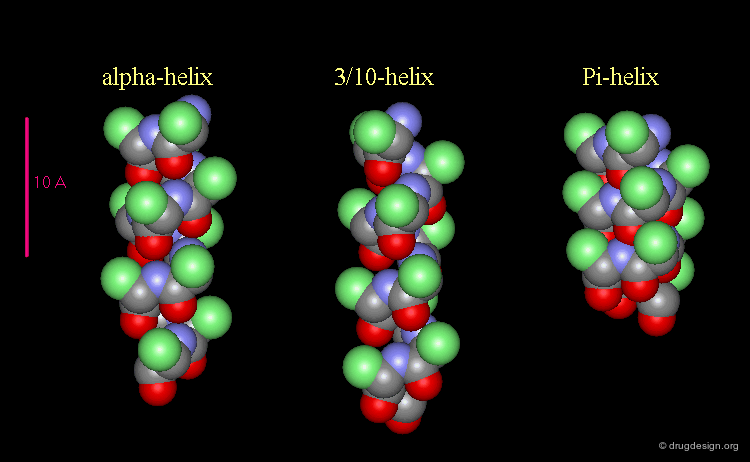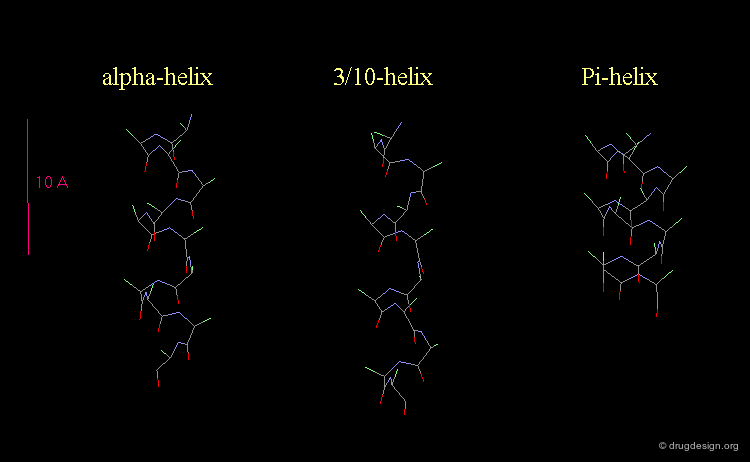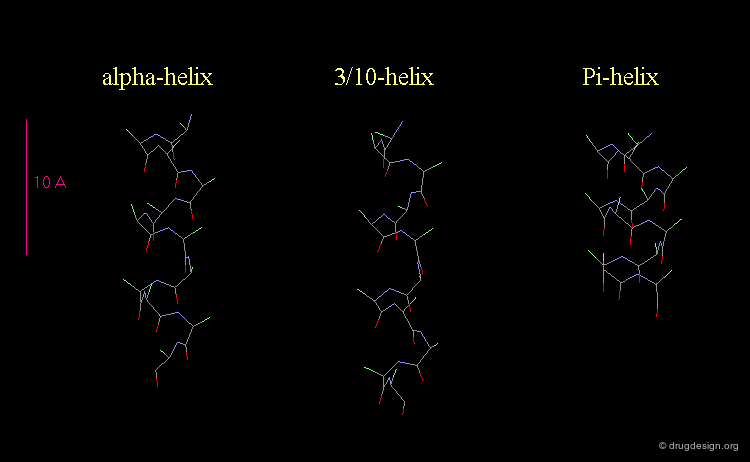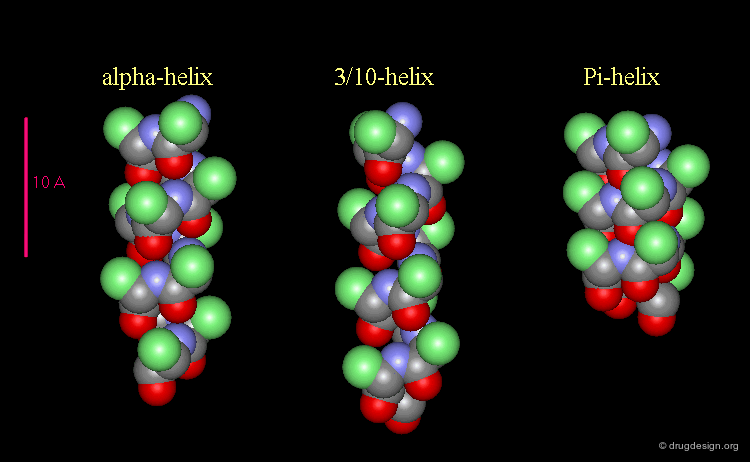
articles
Helix Geometry in Proteins Barlow DJ and Thornton JM J. Mol. Biol. 201 1988
Variants of 3(10)-Helices in Proteins Pal L, Basu G, and Chakrabarti P Proteins 48 2002
Helices Geometrical Parameters¶
The parameters of the three types of helices presented in the previous page are summarized here.
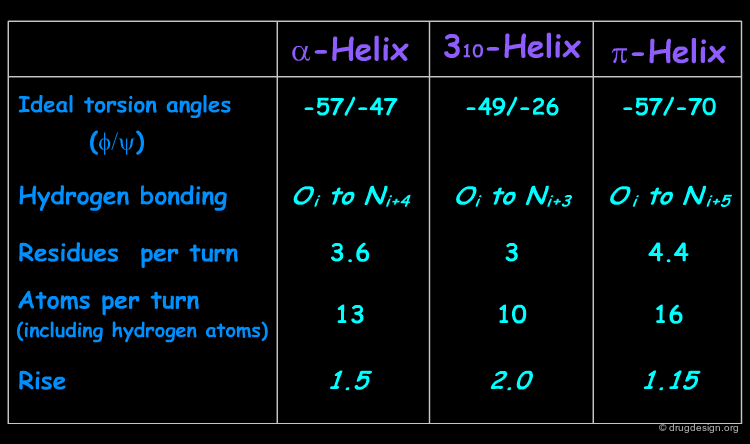

Occurrence of Helices in Proteins¶
The α-helix is found in many proteins. The 310-helix structure is much less widespread and may be found at the beginning or at the end of an α-helix (in a stretch of up to 4 residues). The π-helix appears to be extremely rare and is considered to be unstable.

The β-Sheet¶
The β-pleated sheet or simply "β-sheet" is another major structural element in proteins. As in the α-helix, the structural elements of the β-sheet are held together by hydrogen bonds between N-H and C=O groups of peptide units.
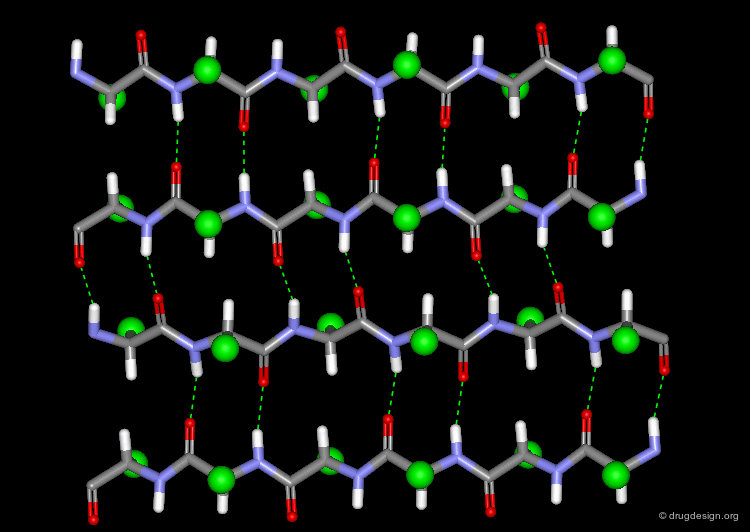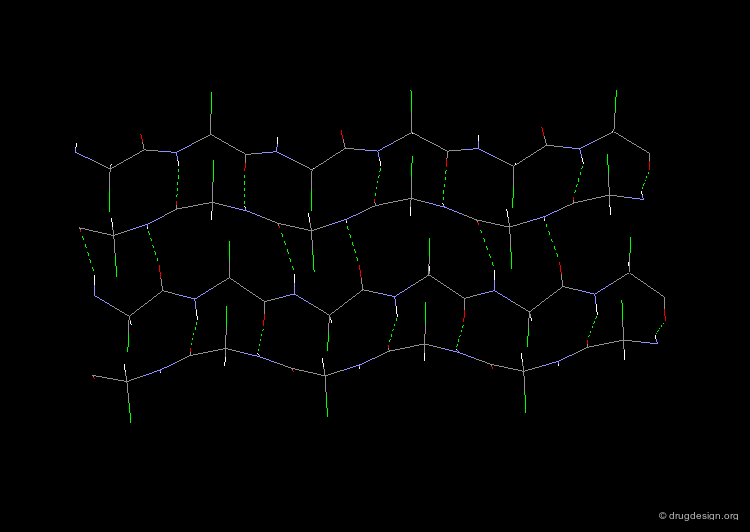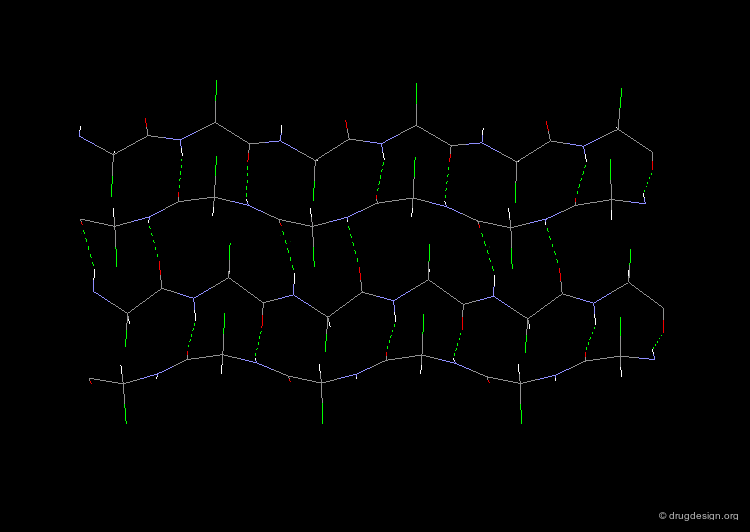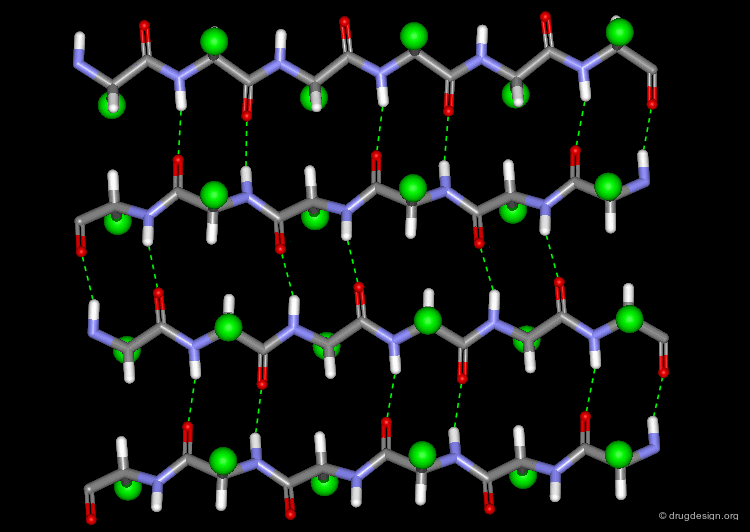
articles
Conformation of Twisted Beta Sheets in Proteins Chothia C J. Mol. Biol. 75 1973
Structure of beta-Sheets. Origin of the Right-Handed Twist and of the Increased Stability of Antiparallel over Parallel Sheets Chou KC, Pottle M, Nemethy G, Ueda Y,and Scheraga HA J. Mol. Biol. 162 1982
Role of Interchain Interactions in the Sabilization of the Right-Handed Twist of beta-Sheets Chou KC, Nemethy G, Scheraga HA J. Mol. Biol. 168 1983
Is the Parallel or Antiparallel beta-Sheet More Stable? A Semiempirical Study Gailer C and Feigel M J. Comput. Aided Mol. Des. 11 1997
Twist and Shear in beta-Sheets and beta-Ribbons Ho BK and Curmi PM J. Mol. Biol. 317 2002
Configurations of Polypeptide Chains with Favored Orientations around Single Bonds: Two New Pleated Sheets Pauling L and Corey RB Proc. Nat. Acad. Sci. USA 37 1951
Beta-Sheet Topology and the Relatedness of Proteins Richardson JS Nature 268 1977
Conformational and Geometrical Properties of Beta-Sheets in Proteins. III. Isotropically Stressed Configurations Salemme FR J. Mol. Biol. 146 1981
Conformational and Geometrical Properties of Beta-Sheets in Proteins. I. Parallel Beta Sheets Salemme FR and Weatherford DW J. Mol. Biol. 146 1981
Conformational and Geometrical Properties of Beta-Sheets in Proteins. II. Antiparallel and Mixed Beta Sheets Salemme FR and Weatherford, DW J. Mol. Biol. 146 1981
Free Energy Determinants of Secondary Structure Formation: II. Antiparallel beta-Sheets Yang AS and Honig B J. Mol. Biol. 252 1995
The Anatomy of Protein beta-Sheet Topology Zhang C and Kim SH J. Mol. Biol. 299 2000
The β-Strand Unit¶
The basic unit of the β-sheet is a single polypeptide chain with an extended, zigzag like conformation, called the β-strand. The side chains (green spheres) of neighboring residues in the β-strand point in opposite directions.
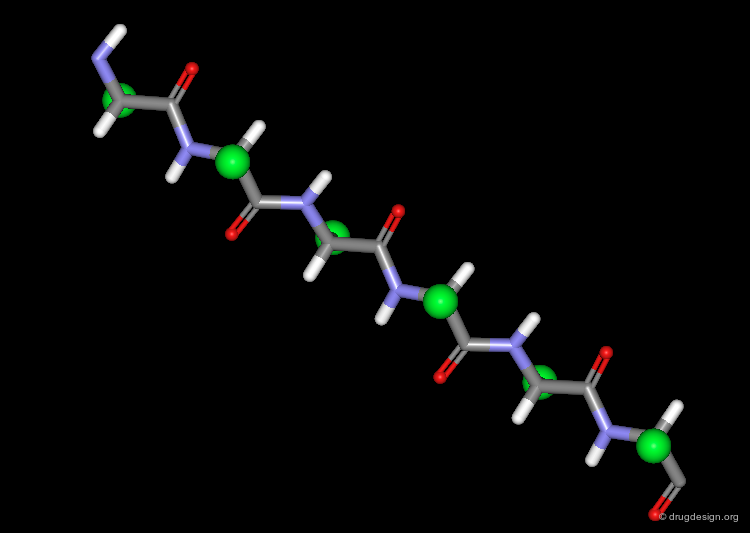
Φ and Ψ Torsion Angles in β-Sheets¶
A relatively broad range of extended structures is sterically possible. Typical values are Ψ=140° and Φ=-130°.

Stability of the β-Sheet¶
Although unstrained, a β-strand alone is not a stable conformation because of the lack of favorable interactions between non-bonded atoms. The strands are much more stable when two or more strands are associated by main chain hydrogen bond interactions, and form a sheet.

Parallel and Anti-Parallel β-Sheets¶
Adjacent strands in a β-sheet can be either parallel (same direction of the polypeptide chain) or anti-parallel (opposite direction of the polypeptide chain). The hydrogen bonding pattern in the two sheets is somewhat different.

Occurrence of β-Sheets in Proteins¶
There is no special preference observed for parallel or anti-parallel β-sheet formation. However, small parallel sheets are relatively rare, probably because of a lower intrinsic stability relative to the anti-parallel form. In a protein, one can find pure parallel or anti-parallel β-sheets, and also a combination of both types.

Twist of the β-sheet¶
The β-sheet model theoretically presented was planar; however most β-sheets observed in proteins are twisted with a twist that is always left-handed (as indicated below). The twist of the sheet results from a torsion in the strand of about 0°-30° per residue.

Turns¶
Another important type of secondary elements is the turn, which is used to reverse the direction of the polypeptide chain. Various types of turns are found in proteins, which differ in the number of residues, the backbone torsion angles, and the hydrogen bond pattern. Here are some examples:

articles
GD. Beta-Turns in Proteins Chou PY and Fasman J. Mol. Biol. 115 1977
Chain Reversals in Proteins Lewis PN, Momany FA, and Scheraga HA Biochem. Biophys. Acta. 303 1973
Loops, Bulges, Turns and Hairpins in proteins Milner-White EJ and Poet R Trends Bioche. Sci. 12 1987
Turns in Peptides and Proteins Rose GD, Gierasch LM and Smith JA Adv. Prot. Chem. 37 1985
Analysis and Prediction of the Different Types of beta-Turn in Proteins Wilmot CM, and Thornton JM J. Mol. Biol 203 1988
Beta Turns and their Distortions: A Proposed New Nomenclature Wilmot CM, and Thornton JM Protein Eng. 3 1990
β-Turns¶
Type I and Type II β-turns are frequently found in protein structures. In these turn types a hydrogen bond is formed between the backbone C=O group and N-H group separated by one residue (i.e. hydrogen bond between residue i and residue i+3 of the turn).

Φ and Ψ Torsion Angles of β Turns¶
Different β-turns have different conformations. The torsion angles for residues i+1 and i+2 in type I and type II β-turns are indicated in the following Ramachandran plot. Note that in a type II β-turn, residue i+2 lies in a region that was allowed only for glycine dipeptides.

Non-Regular Coil and Loops¶
Approximately 80-90% of the residues in proteins can be classified as participating in secondary structures (α-helix, β-sheet or turn). All other residues possess non-regular structures that are described as "coils" or "loops".

articles
Loops in Globular Proteins: A Novel Category of Secondary Structure Leszczynski JF and Rose GD Science 234 1986
Long Loops in Proteins Martin ARC, Toda K, Stirk HJ, and Thornton JM Prot. Eng. 8 1995
book
Hutchinson EG, Morris AL, and Thornton JM Structure Correlation VCH Publishers 1994
Coil¶
A "coil" is the terminology which is used to describe the structure of a polypeptide chain that does not fall into the categories of helix, sheet or turn.

Loops¶
A loop is any stretch of a non-regular polypeptide chain connecting secondary structures. Among the many possible loop conformations, several specific structures have been identified and added to the repertoire of secondary structures. One type of loop-like chain is the Ω-loop which is visualized below (in white).

Super-Secondary Structure (Motifs)¶
Super-Secondary Structures and Motifs¶
Secondary structural elements can be combined to define precise three-dimensional architectures. In many cases it is possible to identify recurring patterns of secondary structure organization in many different proteins, even with completely different sequences. These patterns are called super-secondary structures or structural motifs.
Classification of Super-Secondary Structures¶
There are three main classes of super-secondary structures: all-α, all-β and mixed α/β structures. Simple super-secondary structures consist of 2 to 3 secondary elements. These can be assembled into larger super-secondary structures that are sometimes referred as "folds".

All β super-secondary structures¶
Representative β-super-secondary structures are illustrated in the following pages. The super-secondary structures are represented by both their 3D architecture and by their 2D schematic topological arrangement.

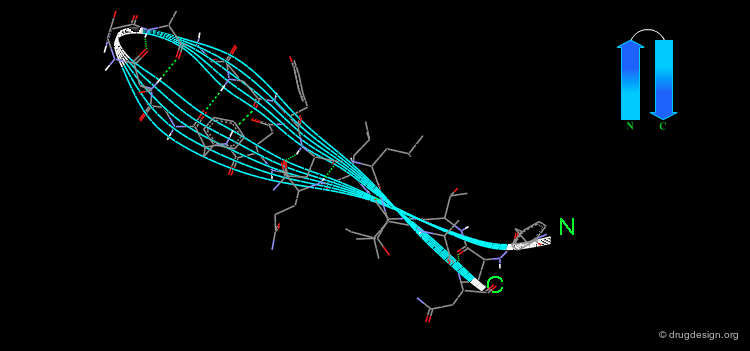
β-Hairpin¶
The "β-hairpin" is one of the simplest and most common super-secondary structures. It includes two consecutive anti-parallel β-strands connected by a loop. The loop itself is referred to as the β-hairpin. β-hairpins adopt a specific conformation which depends on their length and sequence. The length is usually less than 7 residues and often consists of only two residues. The two-residues β-hairpin super-secondary structure adopts one of the classical β-turn conformations.
articles
Structure of beta-beta-Hairpins and beta-beta-Corners Efimov AV FEBS Lett. 284 1991
Beta-Hairpin Families in Globular Proteins Sibanda BL and Thornton JM Nature 316 1985
Conformation of beta Hairpins in Protein Structures Sibanda BL, Blundell TL, and Thornton JM J. Mol. Biol. 206 1989
Beta-Sheet Topology and the Relatedness of Proteins Richardson JS Nature 268 1977
β-Meander¶
β-hairpins can be combined into consecutive anti-parallel β-strands and form the "β-meander" super-secondary structure. Try to find three other β-meander motifs in this 3D structure!

articles
Beta-Sheet Topology and the Relatedness of Proteins Richardson JS Nature 268 1977
Greek-Key¶
Another combination of β-strands is the "Greek-key" super-secondary structure. This is a four-stranded β-sheet motif which is characterized by the topology indicated below. In 3D, this motif can adopt a variety of architectures, either with all four strands in the same β-sheet or in two different sheets, as seen here.

articles
The Greek Key Motif: Extraction, Classification and Analysis Hutchinson EG, and Thornton JM Protein Eng. 6 1993
All α Super-Secondary Structures¶
Representative α-super-secondary structures are illustrated in the following pages. The super-secondary structures are described by both their 2D schematic topological arrangement and their 3D architecture.

αα-Hairpin¶
Helix hairpin or "αα-hairpin" loops connect two sequential α-helices which lie adjacent in space and run approximately anti-parallel. Note the tight packing between the two helices, which is usually achieved by hydrophobic interactions between the side chains.

articles
Structure of alpha-alpha-Hairpins with Short Connections Efimov AV Protein Eng. 4 1991
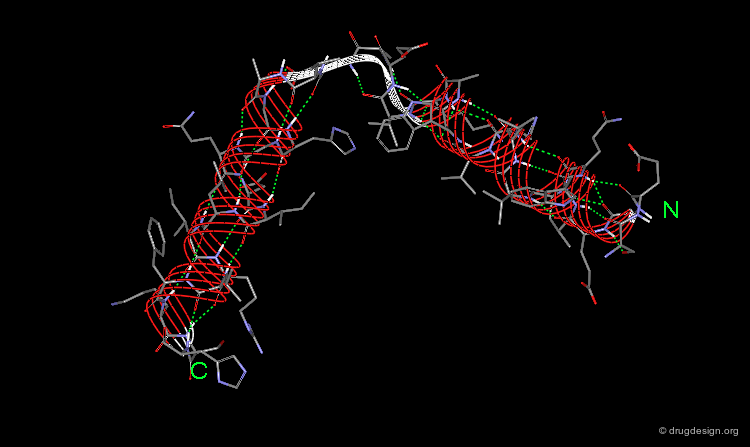
αα-Corners¶
"αα-corners" are short loops which connect helices that are roughly perpendicular. An example is illustrated below (in white).
articles
A Novel Super-Secondary Structure of Proteins and the Relation between the Structure and Amino Acid Sequence Efimov AV FEBS Lett 166 1984
EF Hand¶
Some super-secondary structures have important functions, for example the "EF hand", which is used for calcium binding. This is a view of a helix-loop-helix in which the two helices are roughly perpendicular. The loop is made up of about 12 residues with a conserved sequence.

articles
Calcium Binding and Conformational Response in EF-hand Proteins Ikura, M Trends Biochem. Sci. 21 1996
Carp Muscle Calcium-Binding Portein. II Structure Determination and General Desription Kretsinger RH and Nockolds CE J. Biol. Chem 248 1973
Evolution of the EF-hand Family of Calcium-Binding Proteins Perret C, Lomri N, and Thomasset M Adv. Exp. Med. Biol. 269 1990
Helix-Turn-Helix¶
Another important functional helix-loop-helix super secondary structure is called the "helix-turn-helix" (HTH), although it does not include a true reverse turn geometry. This super secondary structure is used for DNA binding. The HTH is generally composed of two α-helices connected by a short loop of usually 4 conserved residues. The helices are oriented at about 120°. One helix (the yellow helix in the view) is the DNA recognition helix which lies in the DNA major groove.

articles
DNA Recognition by Proteins with the Helix-Turn-Helix Motif Harrison SC and Aggarwal AK Annu. Rev. Biochem 59 1990
Alpha-Helical Protein Assembly Motifs Kohn WD, Mant CT, and Hodges RS J. Biol. Chem. 272 1997
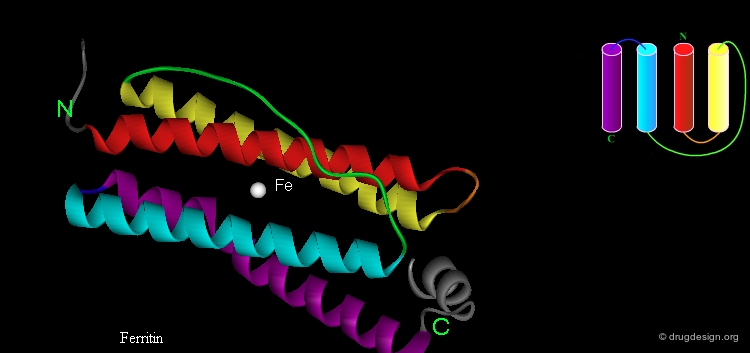
Four-Helix Bundle¶
The "Four-Helix Bundle", in which 4 α-helices are packed together, is another type of common super-secondary structure. This motif has several topologies, as can be seen here. Typically the tight packing of the helices is a result of the amphipathic nature of the helices that puts hydrophobic residues in the interface between the helices and the polar side chains on the outside surface.


articles
Electrostatic Interactions between Loops and alpha-Helices in Four-Helix Bundle Proteins Carlacci L and Chou KC Prot. Eng. 4 1990
Energetics of the Structure of the Four Helix Bundle in Proteins Chou KC, Maggiora GM, Nemethy G, and Scheraga HA Proc .Natl. Acad. Sci. USA 85 1988
Four Helix Bundle Diversity in Globular Proteins Harris N, Presnell SR, and Cohen FE J. Mol. Biol. 236 1994
Alpha-Helical Protein Assembly Motifs Kohn WD, Mant CT, and Hodges RS J. Biol. Chem. 272 1997
The Topological Distribution of Four-alpha-Helix Bundles Presnell SR and Cohen FE Proc. Natl. Acad. Sci. USA 86 1989
Electrostatic Stabilization in Four-Helix Bundle Proteins Robinson CR and Sligar SG Prot. Sci. 2 1993
Principles of Helix-Helix Packing in Proteins: the Helical Lattice Superposition Model Walther D, Eisenhaber F, Argos P J. Mol. Biol. 255 1996
Structural and Functional Diversity in the Four Helix Bundles Weber PC and Salamme FR Nature 287 1980
Mixed α & β Super-Secondary Structures¶
Representative α/β-super-secondary structures are described in the following pages. The super-secondary structures are represented by both their 2D schematic topological arrangement and their 3D architecture.

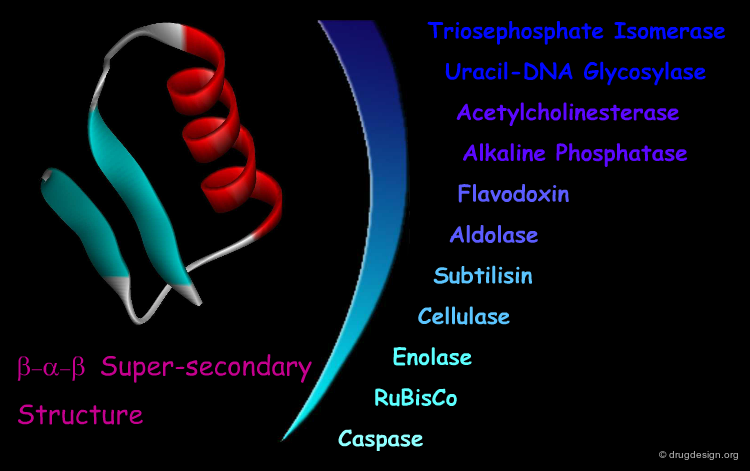
β-α-β Motif¶
The "β-α-β" super-secondary structure consists of a β-strand-loop-α-helix-loop-β-strand. This motif connects two parallel strands and is found in proteins that have parallel β-sheets. The three secondary structure elements are tightly packed and create a hydrophobic core. The loop regions in the motif can vary in length from one residue to more than 100 residues. The β-α-β motif is almost always found with a right-handed fold.

articles
Packing of alpha-Helices onto beta-Pleated Sheets and the Anatomy of alpha/beta Proteins Janin J and Chothia C J. Mol. Biol. 143 1980
Logical Analysis of the Mechanism of Protein Folding. IV. Super-secondary Structures Nagano K J. Mol. Biol. 109 1977
Handedness of Cross Over Connections in beta Sheets Richardson JS Proc. Natl. Acad. Sci. USA 73 1976
On the Conformation of Proteins: the Handedness of the beta-Strand-alpha Helix-beta Strand Unit Sternberg MJE, and Thornton JM J. Mol. Biol. 105 1976
Rossmann Fold¶
An extension of the β-α-β super-secondary structure can lead to the "Rossmann fold" that is often present in nucleotide-binding proteins. This is a β-α-β-α-β motif in which the strands form a parallel β-sheet and the helices lie anti-parallel to the strands on one side of the sheet.
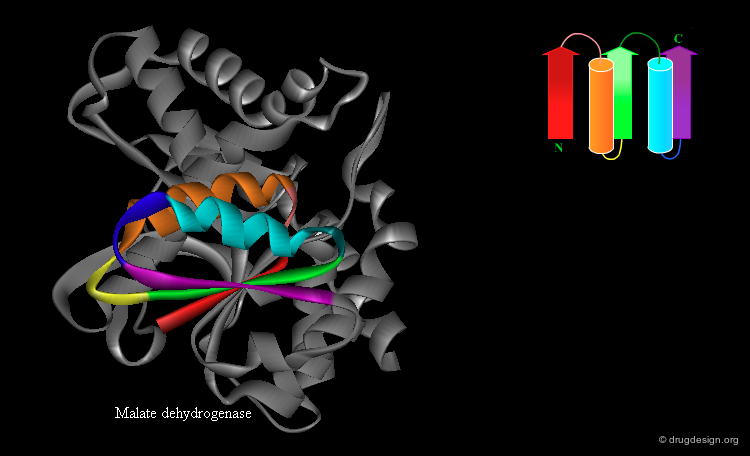
articles
Comparison of Super-Secondary Structures in Proteins Rao ST and Rossmann MG J. Mol. Biol. 76 1973
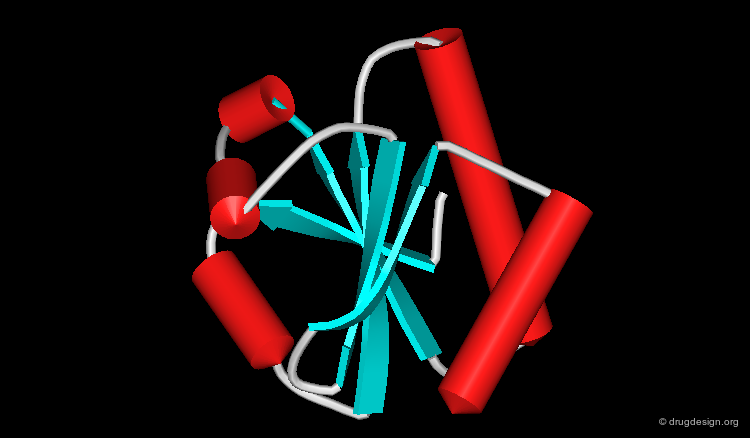
Tertiary Structure¶
Tertiary Structure¶
A combination of all the secondary structure elements and frequently occurring super-secondary structure elements in single polypeptide chains give rise to the tertiary structure of the protein. Thus the tertiary structure refers to the spatial arrangement of all the amino acids atoms in a single polypeptide chain.


Domains in the Tertiary Structure¶
A single polypeptide chain can fold into two or more compact, local and semi-independent structural units which are known as "domains". Domains range in size from about 25 to 500 residues and are frequently connected by only one flexible segment (in light blue).

articles
Parser for Protein Folding Units. Holm L and Sander C Proteins 19 1993
Identification and Analysis of Domains in Proteins Islam SA, Luo J, and Sternberg, MJE Protein Eng. 8 1995
The Anatomy and Taxonomy of Protein Structure Richardson JS Adv. Prot. Chem. 34 1981
Continuous and Discontinous Domains: An Algorithm for the Automatic Generation of Reliable Protein Domain Definitions Siddiqui AS and Barton GJ Protein Sci. 4 1995
A Procedure for Detecting Structural Domains in Proteins Swindells MB Prot. Sci. 4 1995
Domains and Sequence¶
Typically, domains are colinear in sequence, but occasionally a domain will have two or more patches of non sequential segments of the polypeptide chain, and hence will have multiple polypeptide links between domains. By following the chain in this example you can see that the central domain (A) is built of two non-sequential segments (red and pink).

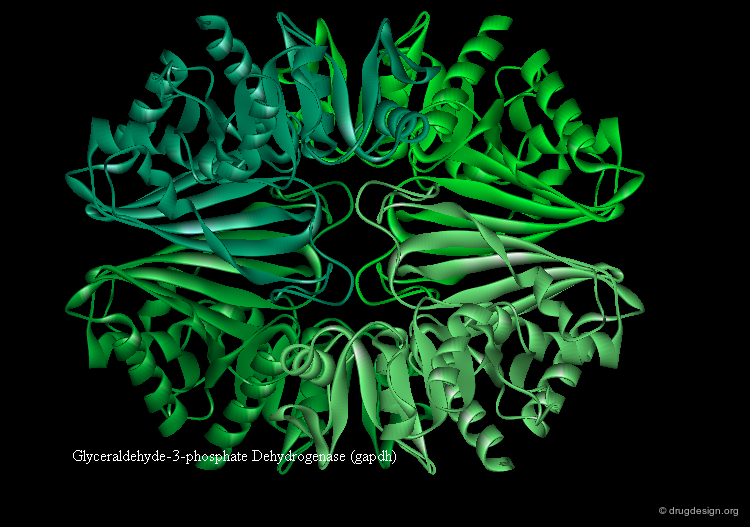
Domains and Function¶
Some proteins have distinct functions for each domain (example gapdh visualized below). In other proteins the function is shared between domains. Ligand binding often occurs in the cleft between two domains (see example fpg).


New Look on Proteins Levels of Architecture¶
The boundary between secondary and tertiary structure is blurred. Super-secondary elements and domains are intermediate levels between secondary and tertiary structures. For this reason super-secondary elements and domains are sometimes considered to be a subset of the tertiary structure and sometimes a distinct level of the protein architecture.




Blurred Boundaries¶
Note that not all helices and strands in a protein necessarily belong to a super-secondary element. Although proteins include secondary and tertiary structures, they do not necessarily include super-secondary elements and domains. Furthermore, some elements (such as the 4-helix bundle) can be considered to be a super-secondary element or domain or the full tertiary structure, depending on the context.



Tertiary Structure Patterns: Folds¶
Each tertiary structure can be defined by its exact fold. Thus, different proteins have the same fold if they have the same major secondary structures in the same spatial arrangement and with the same topological connections.

articles
Biological Meaning, Statistical Significance and Classification of Local Spatial Similarities in Non-homologous Proteins Alexandrov NN and Go N Protein Sci. 3 1994
Catching a Common Fold Blundell TL and Johnson MS Protein Sci. 2 1993
The Classification and Origins of Protein Folding Patterns Chothia C and Finkelstein AV Annu. Rev. Biochem. 59 1990
Identification of Homology in Potein Structure Classification Dietmann S and Holm L Nat. Struct. Biol. 8 2001
Identification of Tertiary Structure Resemblance in Proteins Using a Maximal Common Subgraph Isomorphism Algorithm Grindley HM, Artymiuk PJ, Rice DW, and Willett P J. Mol. Biol. 229 1993
A Database of Protein Structure Families with Common Folding Motifs Holm L, Ouzounis C, Sander C, Tuparev G, and Vriend G Protein Sci. 1 1993
Protein Structure Comparison by Alignment of Distance Matrices Holm L and Sander C J. Mol. Biol. 233 1993
Searching Protein Structure Databases has Come of Age Holm L and Sander C Proteins 19 1994
Mapping the Protein Universe Holm L and Sander C Science 273 1996
New Protein Folds Murzin AG Curr. Opin. Struct. Biol. 4 1994
SCOP: a Structural Classification of Proteins Database for the Investigation of Sequences and Structures. Murzin AG, Brenner SE, Hubbard T, and Chothia C J. Mol. Biol. 247 1995
Identifying and Classifying Protein Fold Families Orengo C, Flores TP, Taylor WR, Thornton JM Protein Eng. 6 1993
CATH- A Hierarchic Classification of Protein Domain Structures Orengo CA, Michie AD, Jones S, Jones DT, Swindells, MB, and Thornton JM Structure 5 1997
Molecular Recognition in Protein Families: a Database of Three-Dimensional Structures of Related Proteins Overington JP, Zhu ZY, Sali A, Johnson MS, Sowdhamini R, Louie C, and Blundell TL Biochem. Soc. Trans. 21 1993
Assigning Genomic Sequences to CATH Pearl FMG, Lee D, Bray JE, Sillitoe I, Todd AE, Harrison AP, Thornton JM, and Orengo CA Nuc. Acids Res. 28 2000
A Data Bank Merging Related Protein Structures and Sequences Pascarella S and Argos P Protein Eng. 5 1992
The Definition of General Topological Equivalence in Protein Structures: a Procedure Involving Comparison of Properties and Relationships through Simulated Annealing and Dynamic Programming Sali A and Blundell TL J. Mol. Biol. 212 1990
Protein Structure Alignment Taylor WR and Orengo CA J. Mol. Biol. 208 1989
Families and the Structural Relatedness among Globular Proteins Yee DP and Dill KA Protein Sci. 2 1993
Fold Diversity¶
From the enormous variability in amino acid sequences found in proteins, one might expect a great diversity of folds, but X-ray crystallography has demonstrated that this is not the case. The tertiary structures of proteins tend to be conserved with frequent occurrences of similar patterns. Many proteins have the same or similar fold, even if there is no obvious evolutionary relation between them.

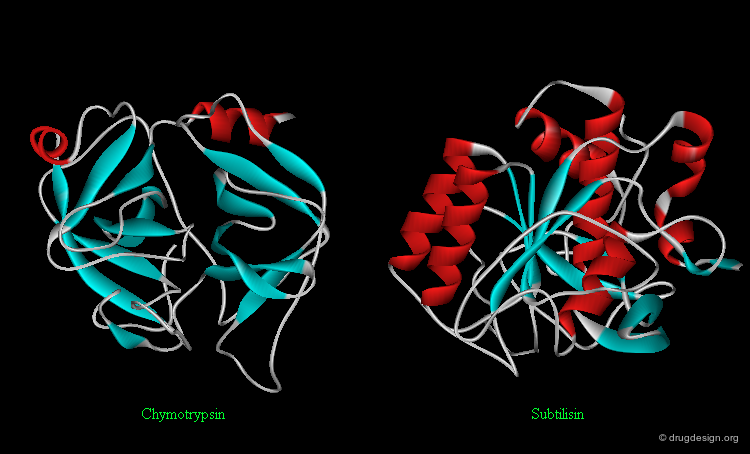
Protein Folds and Function¶
It is important to note that sharing a common fold is not necessarily linked to any functional classification. In general, a certain type of function is not restricted to a certain fold and a certain fold is not restricted to a certain type of function. For example, the following two enzymes (chymotrypsin and subtilisin) are serine proteases: they share the same catalytic mechanism, however they fold in an entirely different way.
Classification of Protein Folds¶
Most of the known proteins and protein domain folds can be classified into three major groups according to their secondary structure content: all (or mainly) α, all (or mainly) β, or mixed α-β folds. Selected examples of the three major fold types are presented in the following pages.

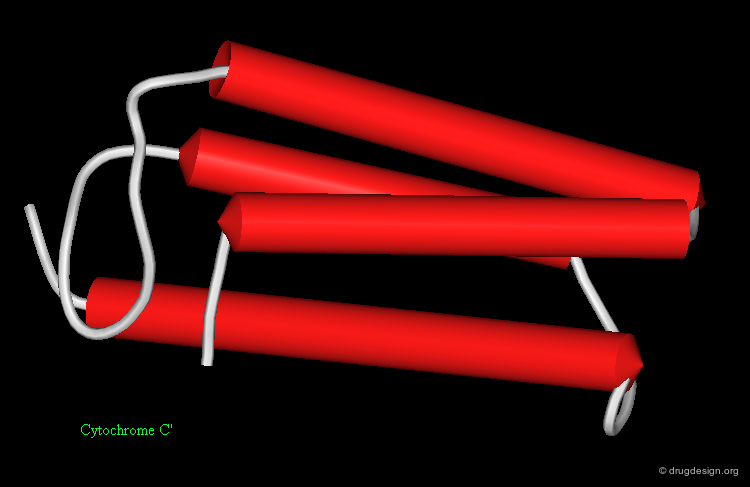
Mainly α Folds¶
Mainly-α-folds include tertiary structures and domains which primarily contain α-helices. Some examples are illustrated here:


articles
Helix to helix packing in proteins Chothia C, Levitt M, and Richardson D J. Mol. Biol. 145 1981
The Packing of alpha-Helices: Simple Coiled Coils Crick FHC Acta Crystallogr 6 1953
Four Helix Bundle Diversity in Globular Proteins Harris NL, Presnell SR, and Cohen FE J. Mol. Biol. 236 1994
General Architecture of the alpha-Helical Globule Murzin AG and Finkelstein AV J. Mol. Biol 204 1988
Structural and Functional Diversity in 4-alpha-Helical Proteins Weber PC and Salemme FR Nature 287 1980
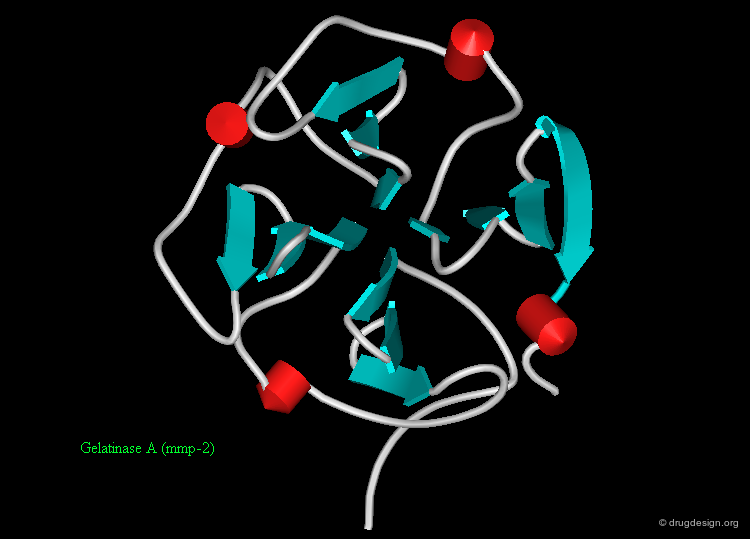
Mainly β Folds¶
Mainly-β-folds include tertiary structures and domains that contain primarily β-strands usually in an anti-parallel arrangement. Some common examples are represented here:



articles
Relative Orientation of Close-Packed beta-Pleated Sheets in Proteins Chothia C and Janin J Proc. Natl. Acad. Sci. USA 78 1981
Orthogonal Packing of beta-Pleated Sheets in Proteins Chothia C and Janin J Biochemistry 21 1982
New Folds for All-beta Proteins Chothia C and Murzin AG Structure 1 1993
Analysis of the Tertiary Structure of Protein beta-Sheet Sandwiches Cohen F, Sternberg MJE, and Taylor WR J. Mol. Biol 148 1981
Structural Principles of Parallel beta-Barrels in Proteins Lasters I, Wodak SJ, Alard P, and Van Cutsem E Proc. Natl. Acad. Sci. USA 85 1988
The Design of Idealized alpha/beta-Barrels: Analysis of beta-Sheet Closure Requirements Lasters, I, Wodak SJ, and Pio F Proteins 7 1990
Structural Principles of alpha/beta Barrel Proteins: the Packing of the Interior of the Sheet Lesk AM, Branden CI, and Chothia C Proteins 5 1989
Gene Duplications in the Structural Evolution of Chymotrypsin McLachlan AD J. Mol. Biol. 128 1979
Structural Principles for the Propeller Assembly of beta-Sheets: the Preference for Seven-fold Symmetry Murzin AG Proteins 14 1992
Beta-Trefoil Fold Murzin AG, Lesk AM, and Chothia C J. Mol. Biol. 223 1992
Principles Determining the Structure of beta-Sheet Barrels in Proteins: I a Theoretical Analysis Murzin AG, Lesk AM, and Chothia C J. Mol. Biol. 236 1994
Principles Determining the Structure of beta-Sheet Barrels in Proteins: II the Observed Structures Murzin AG, Lesk AM, and Chothia C J. Mol. Biol. 236 1994
Twisted Hyperboloid (Strophoid) as a Model of beta-Barrels in Proteins Novotny J, Bruccoleri RE, and Newell J J. Mol. Biol. 177 1984
Conformational and Geometrical Properties of beta-Sheets in Proteins: I Parallel beta-Sheets Salemme FR and Weatherford DW J. Mol. Biol. 146 1981
Conformational and Geometrical Properties of beta-Sheets in Proteins: II Antiparallel and Mixed beta-sheets Salemme FR and Weatherford DW J. Mol. Biol. 146 1981
Conformational and Geometrical Properties of beta-Sheets in Proteins: III Isotropically Stressed Configurations Salemme FR J. Mol. Biol. 146 1981
Mixed α-β Folds¶
Mixed α-β folds include both α-helices and β-strands. This is the most variable class of tertiary structures and domains, some examples are shown here.
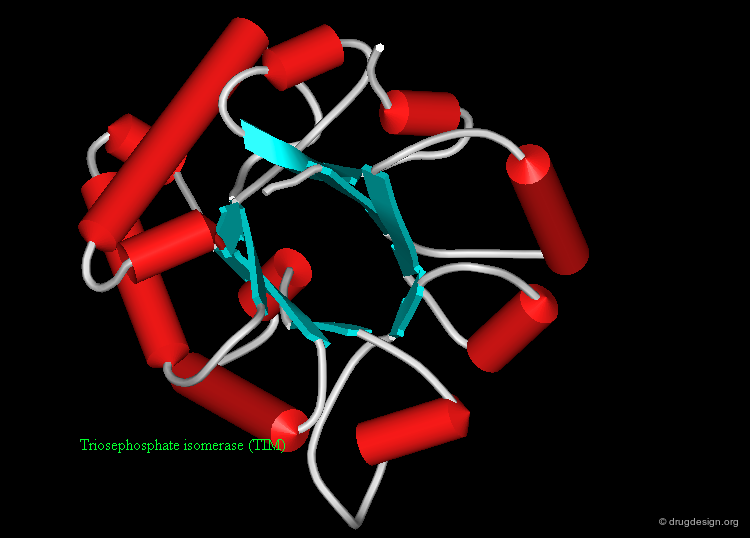


articles
Analysis and Prediction of the Packing of alpha-Helices against a beta-Sheet in the Tertiary Structure of Globular proteins Cohen FE, Sternberg MJE, and Taylor WR J. Mol. Biol. 156 1982
Packing of alpha-Helices onto beta-Pleated Sheets and the Anatomy of alpha/beta Proteins Janin J and Chothia C J. Mol. Biol. 143 1981
Alpha plus Beta Folds Revisited: Some Favoured Motifs Orengo CA and Thornton JM Structure 1 1993
Databases of Folds¶
Each type of class includes several fold types that can be further broken down into subcategories. The hierarchical arrangement of all known protein and proteins domain structures can be found in the SCOP and the CATH databases. FSSP is an additional database of automatic fold classification; however it is a non-hierarchic database.

articles
Mapping the Protein Universe Holm L and Sander C Science 273 1996
SCOP: a Structural Classification of Proteins Database for the Investigation of Sequences and Structures. Murzin AG, Brenner SE, Hubbard T, and Chothia C J. Mol. Biol. 247 1995
CATH- A Hierarchic Classification of Protein Domain Structures Orengo CA, Michie AD, Jones S, Jones DT, Swindells, MB, and Thornton JM Structure 5 1997
Quaternary Structure¶
Quaternary Structure¶
Many proteins, such as the hemoglobin represented here, have more than one polypeptide chain. The different polypeptide chains are called subunits, monomers or protomers. The quaternary structure of a protein refers to the spatial arrangements of its subunits without regard to the internal geometry of the subunit.

Dimers, Trimers, Tetramers etc...¶
The subunit aggregation leads to the formation of protein oligomers which are named for their number of interacting subunits: dimer, trimer, tetramer, etc... The associated subunits can vary from small dimers such as the HIV-1 protease to very large systems such as this chaperon in GroEL tetradecamer.


Homo-Oligomers: Identical Polypeptide Chains¶
The subunits in a protein may be identical. In this case, the resulting homo oligomers are usually symmetrical and almost always exhibit rotational symmetry about one or more axes. Several examples are shown below, according to their symmetry group.



Hetero-Oligomers: Different Polypeptide Chains¶
More than one type of subunit can aggregate together to form the protein unit. In this case it is also common to find multiple copies of various subunits. For example the F1 ATPase has three copies of two different subunits (blue and purple subunits) and three more unique subunits (red, yellow, and green subunits).

Structural Classification of Proteins¶
Structural Classification of Proteins¶
Generally, proteins fall into three main classes, based on their overall 3D structure and their functional role; these are known as Globular, Membrane and Fibrous proteins.

Globular Proteins¶
Most proteins which are found in the aqueous, intracellular environment or in the plasma are globular. They have a somewhat spherical shape or they are made of several compact domains.
Hydrophilic Surface and Hydrophobic Core¶
The globular nature of proteins can be explained by their interactions with the surrounding aqueous solvent. Proteins fold in such a way that most residues with non-polar side chains are buried in the center, creating the protein's hydrophobic core (green side chains), whereas most residues with polar side chains remain exposed on the protein surface (yellow side chains).

Hydrophobic Effect¶
The burial of non-polar residues inside the core of the protein by reducing unfavorable interactions with the surrounding water is known as the "hydrophobic effect". The hydrophobic effect is considered to be one of the most important forces that contributes to the tertiary and also the quaternary structure of globular proteins. The interpretation of this phenomenon is that water-water interactions are the most favorable interactions and in effect, the water squeezes out the hydrophobes so it can interact with itself, leaving the hydrophobes to interact with themselves.

Hydration Layer¶
Globular proteins in aqueous solutions are surrounded by a hydration layer. Fixed water molecules (cyan spheres) occur primarily in positions where they can hydrogen bond to polar groups of the protein.
Membrane Proteins¶
Another class of proteins includes proteins that are attached to biological membranes. This unique class of proteins adapts its structural features according to the extent of interaction with the membrane. The figure describes the characteristic features of the seven transmembrane helix G protein coupled receptors (GPCRs), a family of membrane proteins that cross the cell membrane seven times. They represent an important group of pharmaceutical targets.

articles
Helix Packing in Membrane Proteins Bowie JU J. Mol. Biol. 272 1997
Refinements of the Fluid-Mosaic Model of Membrane Structure Israelachvili J Biochim. Biophys. Acta. 469 1978
Uniformity, Ideality, and Hydrogen Bonds in Transmembrane alpha-Helices Kim S and Cross TA Biophys. J. 83 2002
Mattress Model of Lipid Protein Interactions in Membranes Mouritsen OG and Bloom M Biophys. J. 36 1984
The Prediction of Amphiphilic alpha-Helices Phoenix DA, Harris F, Daman OA, and Wallace J Curr. Protein. Pept. Sci. 3 2002
Hydrophobic Organization of Membrane Proteins Rees DC, DeAntonio L. and Eisenberg D Science 245 1989
The Fluid Mosaic Model of Cell Membranes Singer S and Nicolson, GL Science 172 1972
Membrane Protein Folding and Stability: Physical Principles White SH and Wimley WC Annu. Rev. Biophys. Biomol. Struct. 28 1999
Experimentally Determined Hydrophobicity Scale for Proteins at Membrane Interfaces Wimely WC and White SH Nat. Struc .Biol. 3 1996
book
Gennis R Biomembranes: Molecular Structure and Function Springer-Verlag 1989
Petty H Molecular Biology of Membranes Plenum Press 1993
Sackmann E Handbook of Biological Physics. Vol. 1 Elsevier 1995
Yeagle P The Structure of Biological Membranes CRC Press 1992
The Lipid Bilayer¶
The biological membranes create the cell's physical barriers and compartments, and regulate the molecular traffic across its boundaries. The membrane architecture is the well-known lipid bilayer in which the lipids' hydrophilic polar heads stick out and the hydrophobic tails form the membrane core.


articles
Adhesion Forces of Lipids in a Phospholipid Membrane Studied by Molecular Dynamics Simulations Marrink SJ, Berger O, Tieleman DP, and Jaehnig F Biophys. J. 74 1998
Membrane Model¶
A variety of proteins are linked or embedded in the lipid bilayer and play various roles. Altogether, the lipids and the proteins in the membrane are best described by the dynamic system of the "fluid mosaic model" in which the individual lipid and protein molecules are free to diffuse and interact in the plane of the bilayer.
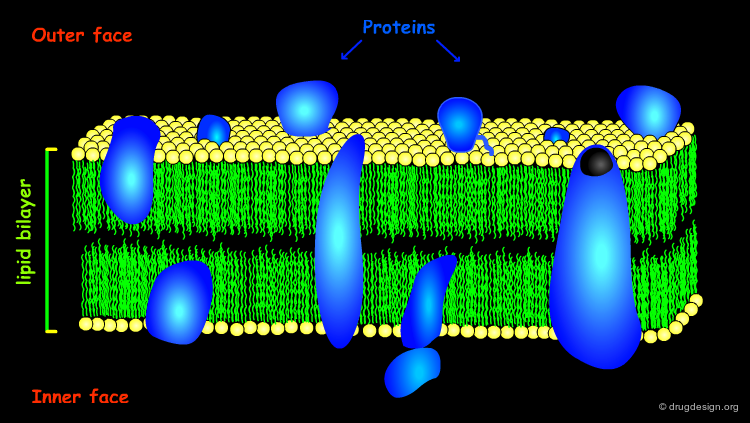
Membrane Proteins Types¶
Different proteins associate with membranes to various extents. Peripheral proteins have a fairly shallow penetration of the membrane surface and possess the same structural features as globular proteins. Integral proteins penetrate deep into the lipid bilayer; these proteins have special structural features that will be discussed in the following pages.

Transmembrane Protein Surface¶
The transmembrane segment of integral membrane proteins must be stable in the hydrophobic hydrocarbon oil-like interior of the lipid bilayer. As a consequence, most of the amino acid side chains that are on the surface of the transmembrane protein must be non-polar (colored in green in the view).
![]()
Transmembrane Protein Folds¶
Furthermore, the polar peptide bond units need to be 'covered' (by creating hydrogen bonds), in order to lower the thermodynamic cost of transferring them to the hydrocarbon interior. This can be achieved either with α-helices (where all peptide bonds are H-bonded internally), or with β-sheet closed structures such as the β-barrel. Currently all transmembrane proteins of known three-dimensional structure correspond to one of these two types.
![]()
![]()
Fibrous Proteins¶
Fibrous proteins tend to be long, thin, insoluble in water and arranged to form fibers by associations side by side to construct macroscopic structures - a feature that is important for their structural roles. Most fibrous proteins have regular, extended structures as we will see in the examples that follow.
Collagen¶
Collagen is the most abundant protein of the human body (it is the matrix protein for bones and skin). Collagen exists as a triple helix in which the individual polypeptide chains helices are very extended (3 Å rise per residue). Each chain is about 1000 residues long (3000 Å in length). The sequence is characterized by Gly residues in every third position, and is also rich in Pro residues with about half of their side chains being hydroxylated. Covalent linking of Lys residues between the polypeptide chains enhances the strength of this structure.

α-Keratin¶
α-keratin is used to form hair, fingernails and skin; it can exist as a dimeric or a trimeric α-helical coiled coil. Each chain is made up of about 300 residues (450 Å in length). Disulfide bonds which are formed between Cys side chains enhance the strength of the structure. The coiled coil structure visualized here comes from another protein, tropomyosin, which is present in the muscles.

Silk Fibroin¶
Silk fibroin is synthesized by spiders for their webs and by silkworms for their cocoons. The architecture of this fibrous protein (made up of β-sheet structures) is illustrated in this theoretical model. The silk fibroin sequence is characterized by specific glycine and alanine rich repeats. Bulky regions with valine and tyrosine residues interrupt the β-sheet stacks and allow for stretchiness.

Perspectives¶
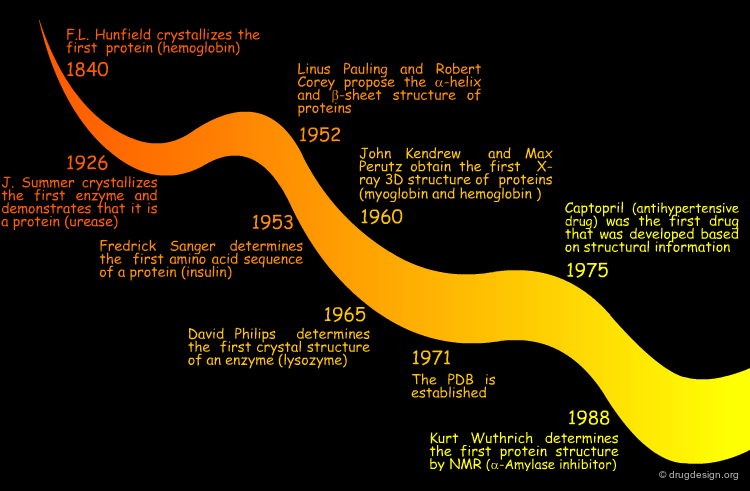
The History¶
Thus little by little, the idea that the biological function of proteins can be understood in terms of their 3D structure has proved to be true. Today, almost half a century after Kendrew and Perutz solved the myoglobin and hemoglobin structures, protein structures have become key features in biology, medicine, and biotechnology.
The Pharmaceutical Connection¶
Protein structures are highly important for the pharmaceutical industry whose role is to convert the knowledge acquired in many areas into useful drugs. 3D structures of disease-relevant proteins are used for the rational design and development of drugs and diagnostic tools. Examples of some protein structures that have served as a basis for the successful design of drugs are shown here (move the mouse to visualize their respective therapeutic areas).

A Fascinating Field¶
This terminates our introduction to Protein Structure. We hope that this chapter has increased your understanding of the complex world of protein structure. The tremendous increase in our knowledge generated by this field, its rapid application and conversion into useful drugs, as well as the beauty of protein structure and function will continue to nurture research focusing on the discovery and application of the subtle machinery underlying life processes. In the years to come progress and advances will doubtless be achieved, far beyond what we can imagine today.
Copyright © 2024 drugdesign.org