Molecular Docking: Principles and Methods¶
Info
Molecular docking consists of the accurate prediction of the orientation and the bioactive conformations of two interacting molecules and the estimation of the tightness of their complex. This chapter presents the docking problem, its basic concepts and the algorithms involved. It includes a presentation of existing docking methods, scoring functions and approaches developed for incorporating flexibility of the interacting molecules. Computational docking is widely used in the pharmaceutical industry and typical examples of applications are presented. A perspective on recent advances is discussed, together with the challenges now being addressed by the scientific community.
Number of Pages: 308 (±7 hours read)
Last Modified: September, 2007
Prerequisites: None
Introduction to Computational Docking¶
Molecular Recognition¶
Molecular recognition is the ability of biomolecules to recognize other biomolecules and selectively interact with them in order to promote fundamental biological events such as transcription, translation, signal transduction, transport, regulation, enzymatic catalysis, viral and bacterial infection and immune response.

articles
Structural basis of macromolecular recognition Wodak SJ, Janin J. Adv Protein Chem. 61 2002 10.1016/S0065-3233(02)61001-0
wikipedia
Molecular Recognition Process: Molecular Docking¶
The molecular recognition process has been nicknamed "Molecular Docking" by analogy to a ship maneuvering in a harbor, because it involves placing molecules in appropriate configurations to interact with a receptor. Molecular Docking is a natural process which occurs within seconds in a cell. In molecular modeling the term refers to the study of how two or more molecular structures fit together.

Understanding Molecular Recognition¶
A good grasp of the principles of molecular recognition at the molecular level and the associated forces involved upon docking is essential to a good understanding of molecular function and biological process. Moreover, knowledge of the mechanical features of a biological signal can be used to design novel therapeutic agents. For example, an inhibitor will be designed to block a recognition process or an agonist will be designed to amplify it.

wikipedia
Molecular Docking Models¶
To get a handle on the molecular recognition process and approach this type of problem better, over the years biochemists have developed numerous models to capture the key elements of the molecular recognition process. Although very simplified, these models have proven highly useful to the scientific community.

The Lock and Key Theory¶
As far back as 1890 Emil Fischer proposed a model called the "lock-and-key model" that explained how biological systems function. A substrate fits into the active site of a macromolecule, just like a key fits into a lock. Biological 'locks' have unique stereochemical features that are necessary to their function.
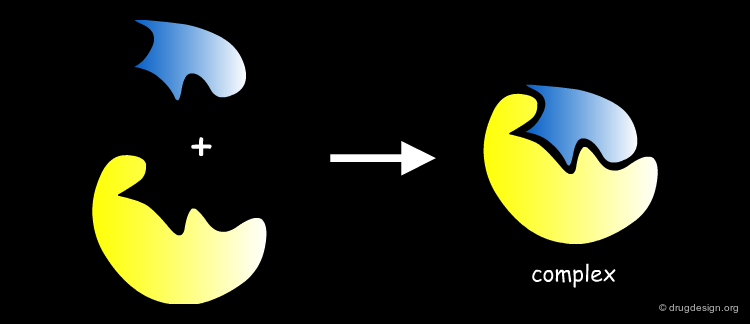
The Induced-Fit Theory¶
In 1958 Daniel Koshland introduced the "induced-fit theory". The basic idea is that in the recognition process, both ligand and target mutually adapt to each other through small conformational changes, until an optimal fit is achieved.


articles
The Key-Lock Theory and the Induced Fit Theory Daniel E. Koshland, Jr. Angew Chem. Int. Ed. Engl. 33 1994
Media
Chey protein Launch protein morphing from momovdb.org
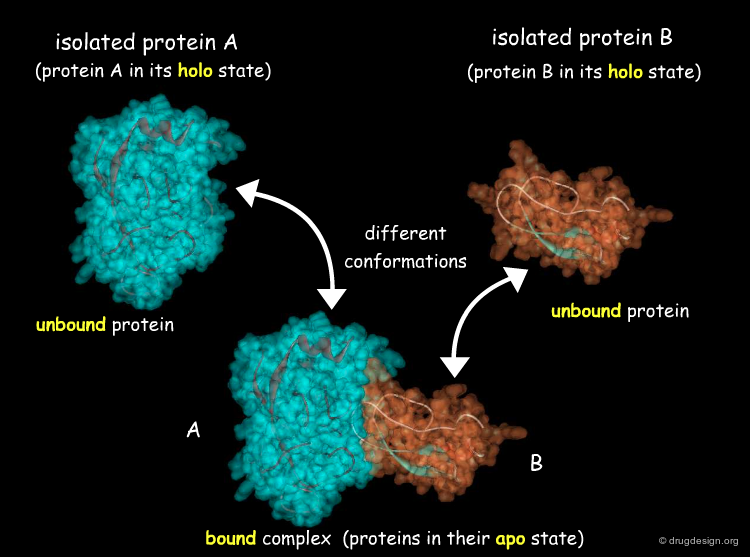
The Conformation Ensemble Model¶
In addition to small induced-fit adaptation, it has been observed that proteins can undergo much larger conformational changes. A recent model describes proteins as a pre-existing ensemble of conformational states. The plasticity of the protein allows it to switch from one state to another.

articles
FOLDING FUNNELS AND BINDING MECHANISMS BUYONG MA ; KUMAR S. ; TSAI C.-J. ; NUSSINOV R. Protein eng. 12(9) 1999 10.1093/protein/12.9.713
Accommodating protein flexibility in computational drug design Carlson HA, McCammon JA. Mol Pharmacol. 57(2) 2000
Molecular Recognition by Induced Fit: How Fit is the Concept? Hans Rudolf Bosshard News Physiol Sci 16 2001
From the Lock and Key to the Ensemble Model¶
The crucial feature to keep in mind is that the lock-and-key, induced-fit and the conformation ensemble model are not contradictory. Each one focuses on a particular aspect of the recognition process. The lock-and-key model introduces the principle of 3D complementarity, the induced-fit model explains how complementarity is achieved, and the ensemble model depicts the conformational complexity of proteins.

Experimental Methods to Study Molecular Docking¶
Numerous experimental techniques are available to study molecular recognition. These include X-ray crystallography, NMR, electron microscopy, site directed mutagenesis, co-immuno-precipitation etc... They allow us to experimentally solve the detailed 3-dimensional structures of biomolecules in their association form which is a necessary step in identifying crucial residues, study the strength of interaction forces, their energetics, understand how molecular structures fit together, and investigate mechanisms of action.
wikipedia
Limitations of Experimental Techniques¶
Despite major advances in structural genomics, experimental techniques are limited by a number of bottlenecks. X-ray crystallography is dependent on the ability to clone, purify and grow a crystal of high enough quality. Solving a structure can take several months. NMR spectroscopy is limited to relatively small proteins (smaller than 25 kDa). Mutagenesis studies are a time-consuming process involving the preparation of a great number of individual mutants.

articles
Prediction of protein-protein interactions by docking methods Smith, G. R. and Sternberg, M. J. E. Current Opinion in Structural Biology 12 2002, 10.1016/S0959-440X(02)00285-3
A Bottleneck in Drug Discovery¶
Due to the limitations of current experimental methods, the initial data available in many drug discovery projects are not generally optimal. The complexes are rarely available in 3D, making a structure-based drug design approach not applicable; a ligand-based approach remains the only alternative for such projects. Note that knowledge of the separated molecules in 3D is only weakly informative if we do not know how to assemble them.

Triggering the Computational Docking Discipline¶
The difficulties in obtaining experimentally structural data of macromolecular complexes have triggered the development of computational predictive methods. Computational docking has emerged as a growing discipline for predicting the way molecules interact and associate together. The process is based on simulation approaches that attempt to find the preferred bound complex of a given system.
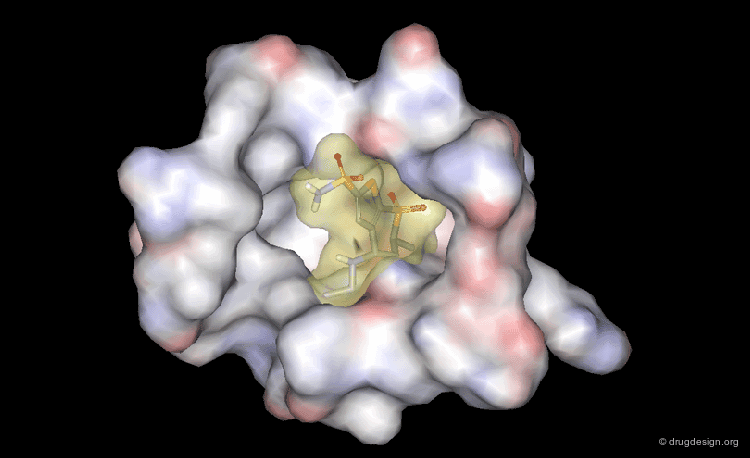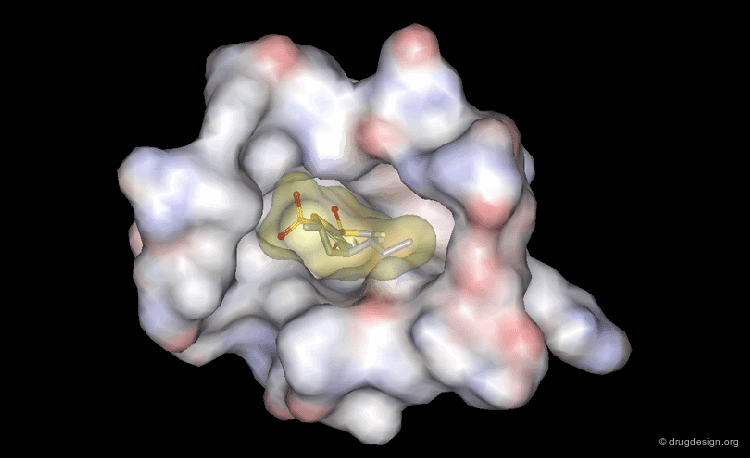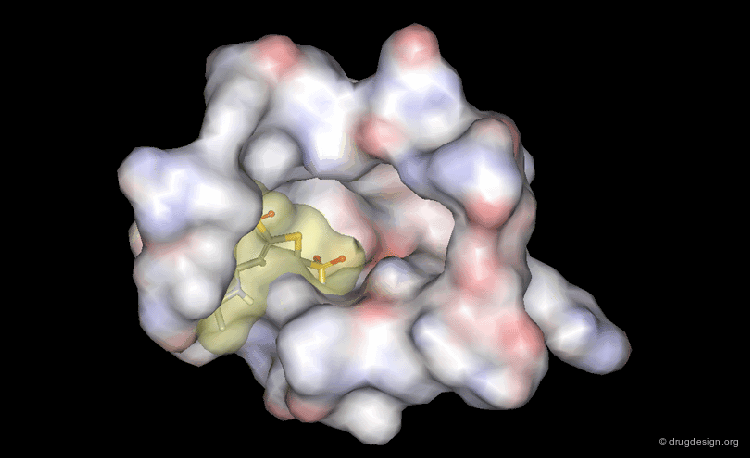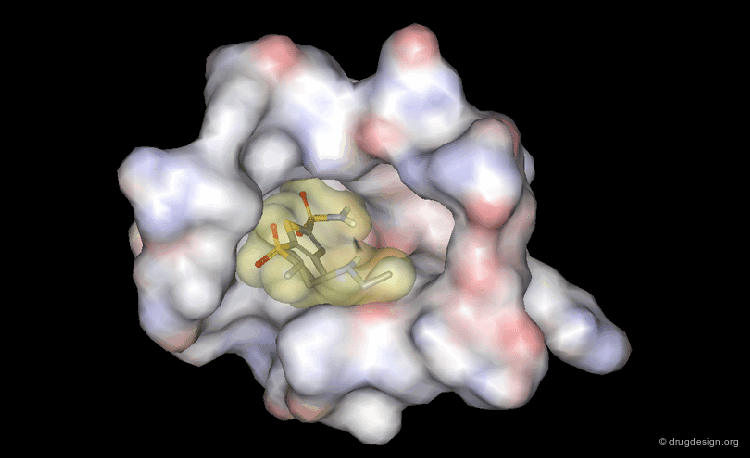
wikipedia
Definition of Computational Docking¶
Computational docking (also called in-silico molecular docking or just docking) is a computational science aiming at predicting the optimal binding orientation and conformation of interacting molecules in space, and to estimate the stability of their complex.

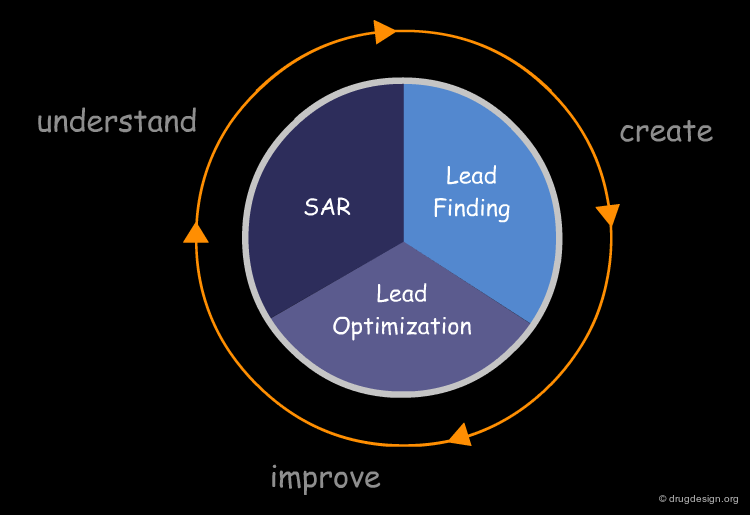
Applications of Computational Docking¶
Computational docking is an essential component in modern drug discovery. Over the last few decades, it has been routinely and successfully applied in most pharmaceutical and biotech companies for a large number of applications. It covers the entire drug discovery field, including structure-activity relationships, lead finding and lead optimization. Some typical applications of molecular docking will be presented in the section entitled "Uses of Docking in Research".
articles
Lead discovery using molecular docking Brian K Shoichet et al. Curr Opin Chem Biol. 6(4) 2002 10.1016/S1367-5931(02)00339-3
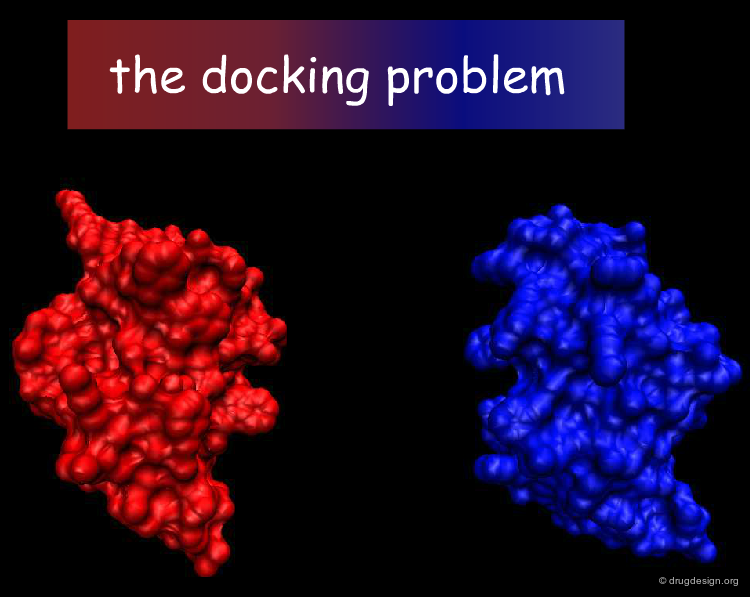
The Docking Problem¶
The Docking Problem¶
The task of predicting how molecules interact with each other is commonly referred to as the 'docking problem'. In this section we define in more detail the terminology used in the field, we present the factors that dictate molecular associations, and explore how the docking problem can be categorized in terms of different difficulty levels, the components of docking softwares and the challenges faced by the scientific community.
Great Diversity of Molecular Interactions¶
The great diversity of types of interactions involved in life processes reflect the diversity of biomolecules associated with biological systems. Molecular docking classifies biomolecules into three categories: small molecules (also called 'ligands'), proteins, and nucleic acids. The most important types of docking systems are: protein-small molecule, protein-protein and nucleic acid-protein interactions. The interactions between a small molecule and a protein are by far much better understood than those between a protein and a nucleic acid.
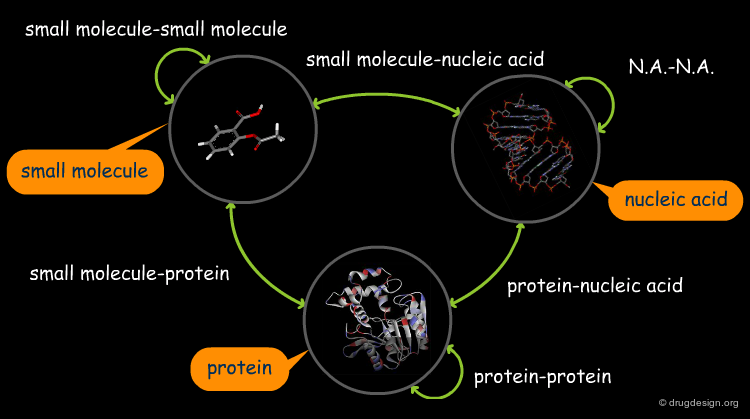
Atomic Basis of Molecular Recognition¶
From a conceptual point of view, molecular recognition is a question of interaction energies between atoms and chemical groups, regardless of whether the atoms belong to a small molecule, a protein or a nucleic acid. Theoretically all docking algorithms could be used for any docking problem. Practically this is not the case, and the huge differences in complexity between docking problems have resulted in algorithms adapted to the particular complexity level that needs to be addressed.

Definition of the "Pose"¶
A "pose" is a term widely adopted for describing the geometry of a particular complex. It refers to a precise configuration which is characterized not only by the relative orientation of the docked molecules but also their respective conformations.

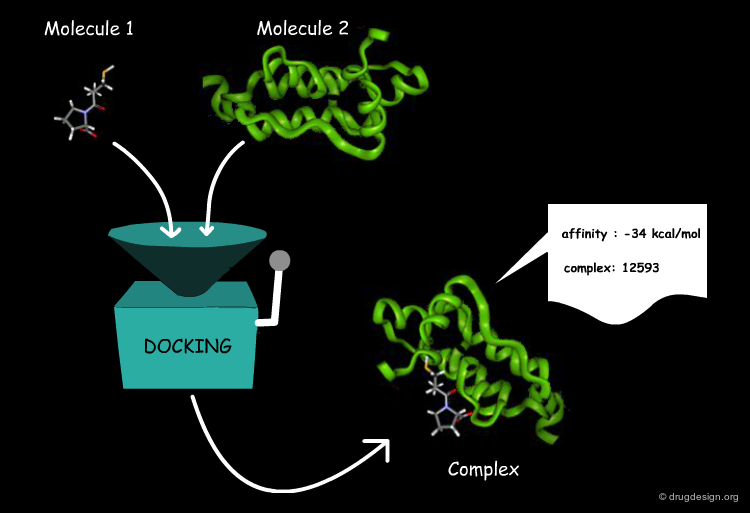
Docking Viewed as a Black Box¶
From the user's point of view, molecular docking software is like a black box whose input is a set of 3D structures to be docked and the output is the predicted complexes for the molecules. More specifically, the user wants to know whether or not the two molecules interact, the binding affinity and the 3D structure of the complex.
Current Computational Docking Programs¶
This chapter describes how difficult computational docking problems are solved in practice. Due to the empirical nature of the algorithms and scoring functions used, current computational docking programs are not always able to predict the correct solution. To compensate for these uncertainties, current software programs propose multiple solutions, consensus results (results obtained using several programs), visual analyses, comparison with known experimental data, point mutation studies etc...

Simulation and non-Simulation Approaches¶
The docking problem can be tackled either using a simulation or a non-simulation approach. Simulation approaches address general methods aiming at reproducing the behavior of any molecular system; when applied to molecular recognition they simulate the docking process. Non-simulation approaches are not meant to analyze the entire process developed in the course of molecular recognition; rather they focus on the last step when the molecules involved finally fit together.

Simulation Approaches¶
Simulation approaches emerged in the 1970s, molecular dynamics (MD) being one of the most developed methods of this type. It applies the laws of classical mechanics to simulate the motion of molecules and their conformational modifications, by computing changes in atomic coordinates as a function of time. To simulate the binding pathway the process starts when the two interacting molecules are separated by some physical distance. At each step, the position and the velocities of the atoms are computed using Newton's equation of motion. Molecular dynamic simulation is presented in some detail in the "Molecular Dynamics" chapter.

articles
Molecular dynamics simulations of biomolecules M. Karplus and A. McCammon Nature Structural Biology 9 2002 10.1038/nsb0902-646
Non-Simulation Approaches¶
Non-simulation approaches do not try to understand the dynamic processes involved in the formation of complexes, rather they aim at answering the following question: assuming that the two molecules do interact, which relative position corresponds to the best interaction? The first non-simulation approach that tackled the docking problem was the DOCK algorithm introduced by Kuntz et al in 1982. This chapter will focus on the non-simulation based approaches.
Molecular Complementarity in Computational Docking¶
All non-simulation approaches used in computational docking exploit the concept of molecular complementarity. The structures interact like a hand in a glove, where both the shape and the physico-chemical properties of the structures contribute to the fit. In the following pages we provide more detail on the concept of complementarity with its underlying structural and energetical content.

articles
MOLECULAR RECOGNITION AND DOCKING ALGORITHMS Natasja Brooijmans, Irwin D. Kuntz Annual Review of Biophysics and Biomolecular Structure 32 2003 10.1146/annurev.biophys.32.110601.142532
Shape Complementarity¶
Shape complementarity is the primary criterion for evaluating the fit in the computational docking of two candidate structures. The intermolecular volume and areas between docked molecules are useful for measuring the degree of shape complementarity. Although anyone can put together two jigsaw-puzzle pieces in seconds, the assembly of molecules is not as easy because of the fuzziness in the 3D surfaces to be fit, and also the sense of incompleteness in the 3D complementarity.

Chemical Complementarity¶
In addition to shape compatibility, chemical and physico-chemical complementarity are also important criteria in the docking between candidate structures. Later on in this chapter we explain how to calculate and assess the complementarity of docked structures.
book
Rarey, M.; Kramer, B.; T., Lengauer 3rd Int. Conf. on Intelligent Systems for Molecular Biology (ISMB'95) AAAI Press 1995
Energy Dictates Molecular Associations¶
What is the real origin of "self-assembly" for shape and chemical complementarity? Docking is not the result of a random process; rather it is driven by a subtle mechanism enabling biological machinery to function. This process is dictated by forces that are energy based: the complex has a lower potential energy than its constituent parts, and this keeps the different parts together.

articles
Toward complex matter: supramolecular chemistry and self-organization. Lehn JM Proc Natl Acad Sci U S A. Apr 16;99(8) 2002 10.1073/pnas.072065599
Supramolecular chemistry Lehn JM. Science Jun 18;260(5115) 1993
Toward self-organization and complex matter Lehn JM. Science Mar 29;295(5564) 2002 10.1126/science.1071063
book
Lehn, J.M.
Weinheim: Wiley-VCH 1995
Find a Complex that Minimizes the Energy¶
Another way to define the goal of computational docking is to find the 3D configuration of the complex that minimizes the energy. The energy is measured by a "scoring function" that accounts for shape and physico-chemical complementarity. Scoring functions will be presented later in this chapter.

Accounting for Molecular Flexibility in Docking¶
The mutual adaptation of a ligand with its receptor as introduced by the induced-fit theory is crucial to understanding ligand binding and protein function. One of the major challenges in molecular docking is how to account for this adaptation in docking calculations.
Flexible Docking: Increasing Levels of Complexity¶
The docking problem can be classified according to the way flexibility is modeled. In ascending order of complexity: (1) ignoring the flexibility of the molecules and treating them like rigid objects; (2) only the ligand is treated as flexible; (3) only the protein is treated as flexible and (4) both protein and ligand are treated as flexible.

Initial Data and Nature of the Docking Difficulty¶
Another way to classify the docking problem is to consider the nature of the initial structural data available. Docking can address three different situations: (1) when an experimental 3D structure of the complex is available, this problem is referred to as "bound docking"; (2) when the 3D structures of the separated molecules are known, this is referred to as "unbound docking"; (3) when no 3D structural information is available, this is referred to as "modeled docking". These situations correspond to increasing levels of difficulty.

articles
Principles of docking: An overview of search algorithms and a guide to scoring functions Halperin I, Ma B, Wolfson H, Nussinov R. Proteins: Structure Function and Genetics Jun 1;47(4) 2002 10.1002/prot.10115
Bound Docking¶
"Bound docking" is the simplest computational docking treatment; its aims at a computerized reconstruction of known experimental complexes. In practice a complex is extracted from the protein databank, and then the molecules are separated. The software tries to reproduce the experimental results by considering the two molecules to be rigid objects. Of minor utility in drug discovery, bound docking is useful for the calibration of docking programs. A heavy atom root-mean-square deviation (RMSD) of less than 2 Å between the experimental structure and the top-ranked pose is often considered to be successful docking. Bound docking algorithms will be presented in the section entitled "Rigid Docking Methods".

articles
A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance Perola E, Walters WP, Charifson PS Proteins: Structure Function and Genetics 56 2004 10.1002/prot.20088
Unbound Docking¶
"Unbound Docking" is a more difficult problem and aims at predicting the complex formation based on experimentally solved structures of the two separated molecules, when the 3D structure of the complex is not known. The challenge of unbound docking lies in our capacity to correctly model protein flexibility into docking calculations. The incorporation of protein mobility into docking is a complex issue that will presented in the section entitled "Methods for Incorporating Flexibility".

Modeled Docking¶
When structural experimental data of the target proteins are not available at all, "Modeled Docking" relies on 3D models constructed by modeling (e.g. homology modeling). This is considered one of the most challenging issues in the development of computational docking. A major concern is how to deal with flexibility when such uncertain approximations are used.

The Three Generations in Computational Docking¶
Computational docking is a recent and rapidly developing technology. Its classification into three types of problems (bound, unbound and modeled docking) corresponds to three successive generations of methods. Given the current state of the art, bound docking has now become mature, and attention is now directed to unbound docking. Predictive modeled docking is still in its infancy.

Three Components of Docking Software¶
Three essential components are generally required in docking software: first, a way to represent structures and properties; second, a method to assess the quality of docked complexes; and finally, an efficient search algorithm that decides which poses to generate. Very often the search and the scoring components are coupled together in an iterative fashion.

articles
Prediction of protein-protein interactions by docking methods Smith, G. R. and Sternberg, M. J. E. Cur ent Opinion in. Structural Biology 12 2002, 10.1016/S0959-440X(02)00285-3
System Representation¶
Molecular Representation¶
Selecting how molecules are represented is a crucial step for all docking algorithms. There are three representations commonly used in docking programs: the atomic representation, the surface representation and the grid representation. The choice of representation dictates the way the docking problem will be tackled. These three representations will be briefly introduced in this section.

Atomic Representation¶
The first and simplest representation of a system is the atomic representation. Each atom is characterized by its coordinates and atom-type. Since the interactions between proteins involve exposed residues, buried residues in this type of representation can be removed.

Complexity of the Atomic Repesentation¶
The atomic representation is generally associated with a list of pair-wise atoms whose interactions need to be evaluated by the scoring function. The huge number of details generated by the atomic representation of biomolecules makes this method computationally very expensive. Some approaches use a simplified representation to reduce this computational complexity. For example two atoms (a carbon alpha and the side chain centroid) can be used to represent a whole residue.

Internal Coordinates¶
Internal coordinates define the positions of the atoms in terms of bond lengths, bond angles and dihedral angles. This representation is preferred to Cartesian coordinates because it enables the easy control of flexibility and key geometrical parameters (such as torsion angles) that can be assigned to particular values or blocked during a whole simulation.

articles
ICM: a new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation Abagyan, R.; Totrov, M.; Kuznetsov, D. J. Comput. Chem. 15(5) 1994 10.1002/jcc.540150503
Prodock: Software package for protein modeling and docking Trosset, J.; Scheraga, H. J. Comput. Chem. 20 1999
Protein Preparation¶
The preparation of the protein calls for great care. Important decisions include the choice of the tautomeric forms of histidine residues, the protonation states of amino-acids and torsion angles of some residues; their incorrect assignments may lead to docking errors. An energy minimization is often needed to remove steric clashes from the initial structure.

Small Molecule Preparation¶
Before generating and docking the 3D structures of a library of ligands, it is important to "clean up" the 2D structures being used by removing any counter ions, salts, or water molecules that might be part of the registered structure. All reactive or otherwise undesirable compounds must also be removed. Possibly generate all stereo isomers, tautomers, and protonation states of the structures. For most docking programs the tautomeric and protonation state of the ligands to be docked is defined by the user; in general the structure considered to be dominant at a neutral pH is generated; here also, incorrect assignments may lead to docking errors.



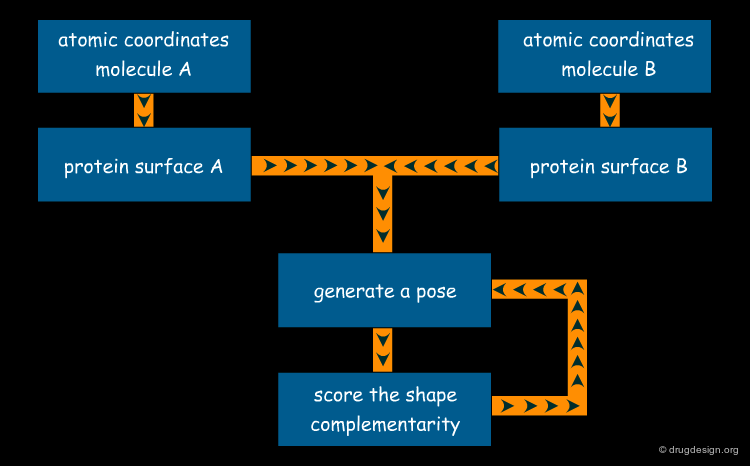
Surface Representation¶
As 3D complementarity (shape and physico-chemical properties) is crucial in the recognition process, many docking algorithms use a surface representation of the system. In particular, protein-protein docking is generally tackled using a surface-based representation. In the following pages the notion of molecular surface will be revisited and several methods for generating and comparing molecular shapes will be presented.
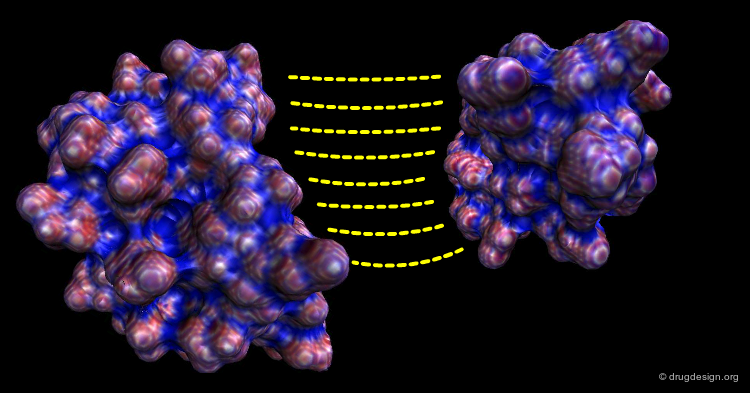
Molecular Surface Matching¶
Shape complementarity between molecules means that there is a good complementarity between their molecular contact surfaces. The atoms present in the interior of the molecules are not directly involved in this complementarity; however they induce particular physico-chemical properties on the molecular surface that add a new dimension to the geometrical complementarity between the surfaces themselves.
Surface-Based Representation¶
Molecules can be represented by their surface or preferably by their so-called "solvent accessible surface". The solvent accessible surface is a kind of expanded van der Waals surface. It corresponds to the molecular envelope of the surface of each atom which is accessible to the center of a probe sphere of a given radius, as explained in the next page.

Accessible Surface Area¶
The accessible surface area of a molecule corresponds to the surface area that is accessible to a probe sphere. It is defined as the locus of the center of a probe sphere as it rolls over the van der Waals surface of the molecule. Small probe radii detect more surface details, whereas increasing the radius progressively smoothes the accessible surface. A typical value for the radius of the probe is 1.4 Å, which approximates the radius of a water molecule (in this case the surface is called the "solvent accessible surface area").

wikipedia
Solvent Contact & Reentrant Surfaces¶
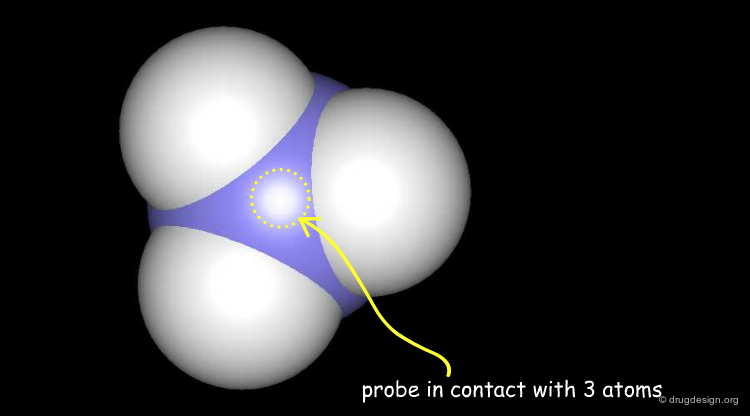
The solvent contact and reentrant surfaces are the most commonly used molecular surface representations. The probe ball is rolled over the molecule, creating three types of surfaces: (1) convex contact surfaces (green points; they belong to one atom); (2) toroidal reentrant surfaces (pink points; they lie between two atoms) and (3) concave reentrant surfaces (blue points; they belong to the patches where the probe is in contact with three atoms). The main advantage of contact and reentrant surfaces is that they model the shape complementarity at the interfaces of interacting molecules very well.
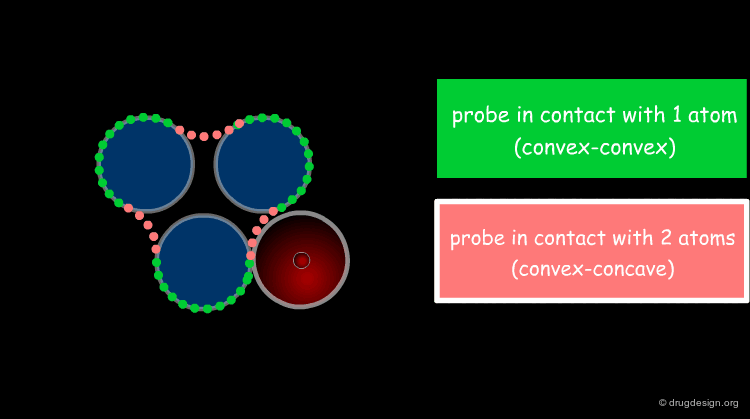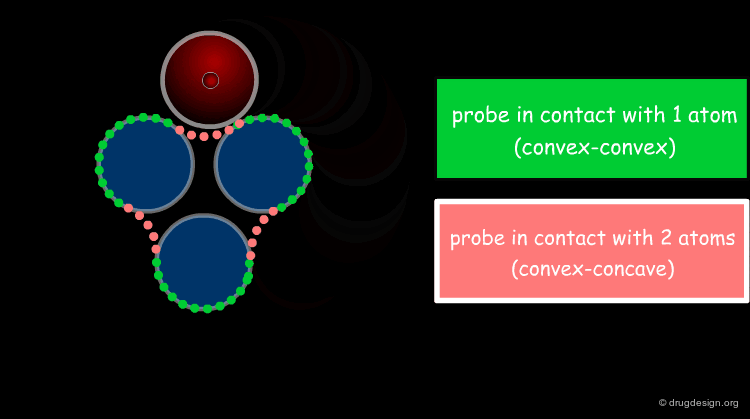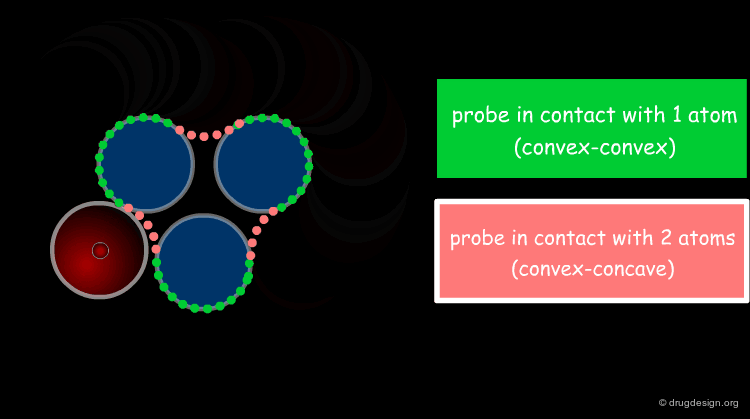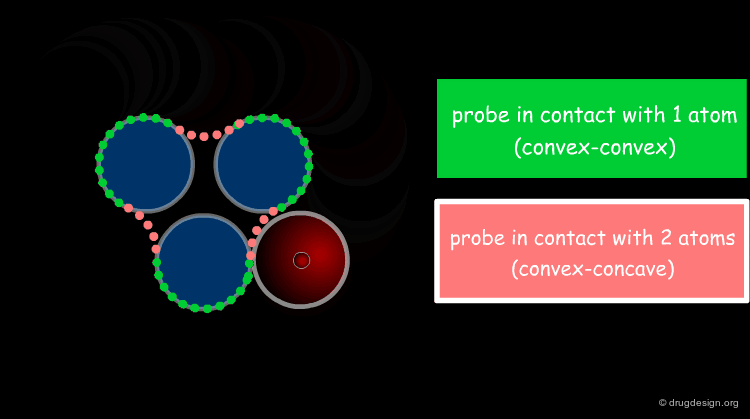
articles
Areas, volumes, packing and protein structure Richards, F Ann. Rev. Biophys. Bioeng. 6 1977
Example of Contact & Reentrant Surface¶
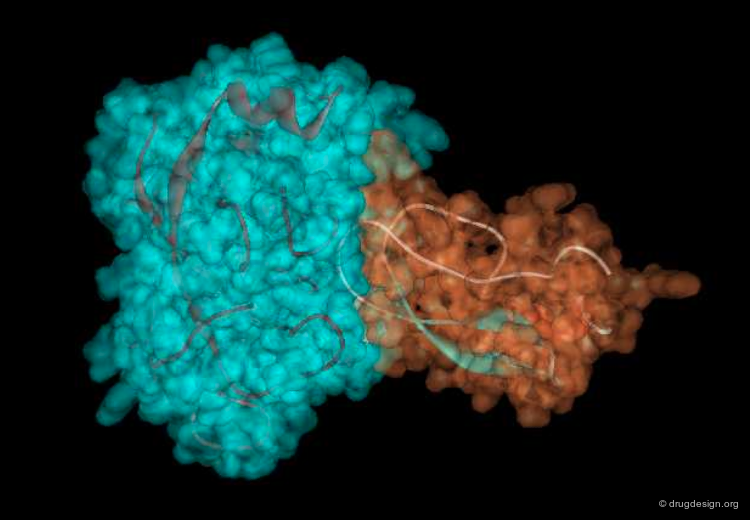
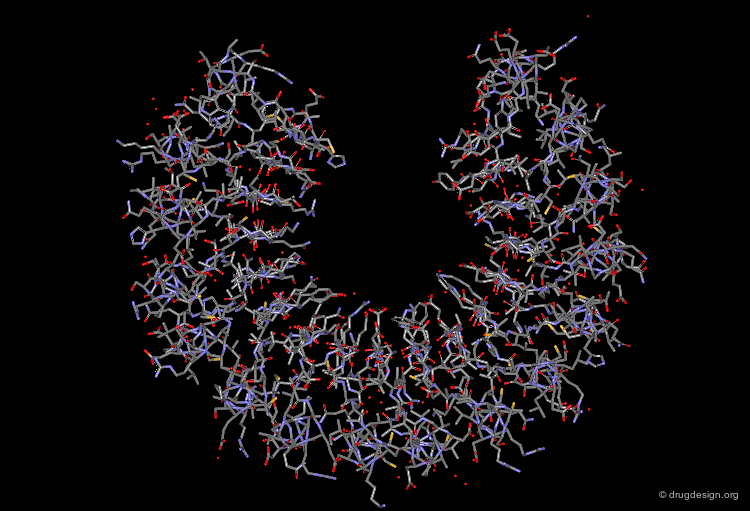
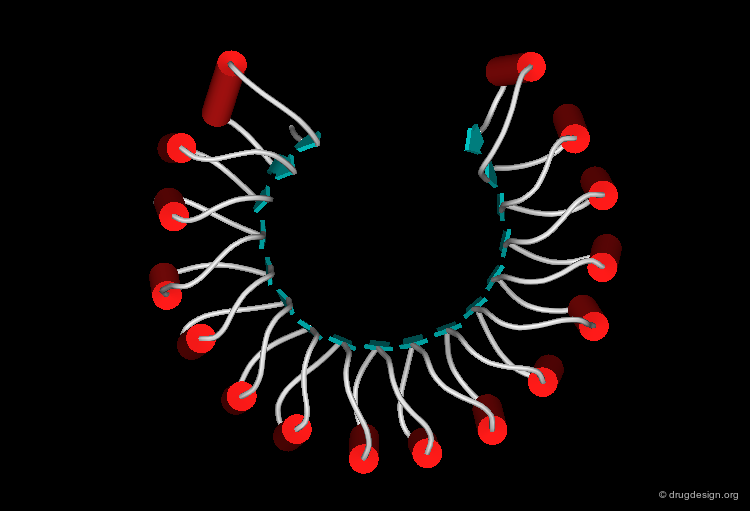
The following view illustrates the solvent contact and reentrant surfaces of ribonuclease inhibitor. Convex contact surfaces are shown in green; toroidal reentrant surfaces in red and concave reentrant surfaces in yellow.

wikipedia
Describing the Molecular Shape¶
Assessing shape complementarity relies on our ability to compare the molecular shapes of interacting molecules. A shape function (or shape descriptors) can be calculated for each molecule and makes this comparison possible. Shape functions are based on critical points which are selected on the molecular surface; they represent points carrying the relevant information. The complexity of the algorithm increases with the number of points.

Connolly's Contact and Reentrant Surfaces¶
In 1983 Connolly introduced his Molecular Surface (MS) program that computes contact and reentrant surfaces. The surfaces are represented by critical points created by placing a water probe tangent to one atom for the contact surfaces (green), and tangent to two (red) or three atoms (yellow) for reentrant surfaces. This implementation paved the way for many docking algorithms based on surfaces.

articles
Solvent-accessible surfaces of proteins and nucleic acids Connolly, M. Science 221 1983 10.1126/science.6879170
Analytical molecular surface calculation Connolly M. J. Appl. Cryst. 16 1983 10.1107/S0021889883010985
Sparse Surface¶
The MS program produces a good surface representation, but the number of points is too large for fast docking algorithms. In 1994 Lin et al. introduced a surface representation by "sparse surface points". Each face of the surface (convex, concave and toroidal) is represented by one point alone. The centroid of each face is computed and then projected onto the surface in the direction of the normal. The projected centroid is a critical point.


articles
Molecular surface representations by sparse critical points Lin, S.; Nussinov, R.; Fischer, D. Wolfson, H. Proteins: Structure, Function, and Genetics 18 1994 10.1002/prot.340180111
Delaunay Triangulation¶
Another way of describing a molecular surface is to construct triangles on it. Delaunay triangulation permits replacing the surface of a protein by a set of triangles. The triangles are constructed in such a way that if a circle were drawn that contained the three points of the triangle, no other points in the set would fall within that circle. Delaunay triangulations can be built easily and rapidly.


wikipedia
"Knob" and "Hole" Descriptors¶
Knobs and holes are examples of shape descriptors used in molecular matching. A method for measuring local curvature was introduced by Connolly: a sphere of radius R is placed at a surface point. If more than 2/3 of the sphere is inside the molecule, the surface point is called a "hole", if less than 1/3 of the sphere is inside the molecule, the point is a "knob".

articles
Shape complementarity at the hemoglobin a1b1 subunit interface Connolly, M. Biopolymers 25 1986 10.1002/bip.360250705
Shape complementarity at protein-protein interfaces Norel, R.; Lin, S.; Wolfson, H.; Nussinov, R. Biopolymers 34 1994 10.1002/bip.360340711
Using Knobs and Holes for Complementarity¶
The shape complementarity of molecular surfaces in a complex is quantified by the shape function: a knob of one molecule should match with a hole of the other molecule. Knobs and holes are used as complementary features by a number of docking algorithms.

Other Examples of Shape Descriptors¶
In addition to the simple concept of knobs and holes, other shape descriptors have been introduced for assessing the geometrical complementarity between two molecular surfaces. This includes the quadratic shape descriptor (QSD), surface patches, molecular surface cubes, and surface normals.
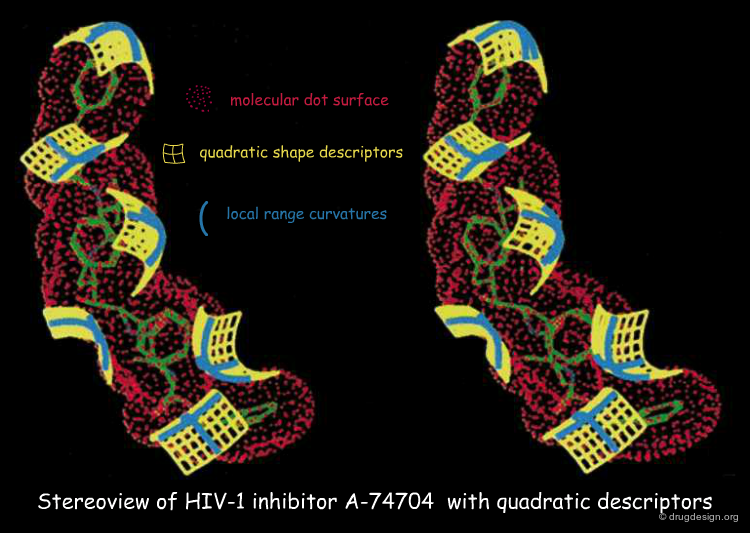
articles
Quadratic Shape Descriptors. 1. Rapid Superposition of Dissimilar Molecules Using Geometrically Invariant Surface Descriptors. Goldman BB, Wipke WT. J.Chem. Inf. Comput. Sci. 40(3) 2000 10.1021/ci980213w
QSD quadratic shape descriptors. 2. Molecular docking using quadratic shape descriptors (QSDock) Goldman BB, Wipke WT. Proteins 38(1) 2000 10.1002/(SICI)1097-0134(20000101)38:1<79::AID-PROT9>3.0.CO;2-U
wikipedia
Grid Representation¶
The third representation is the grid representation pioneered by Goodford in 1985. It was introduced in docking algorithms by Katchalski-Katzir et al. and Jiang and Kim in the early 1990s. The idea is to replace the protein by a grid where the interaction energies between the protein and a chemical probe (typically Van der Waals and electrostatic interactions) at each grid point are calculated and stored.
articles
Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques Katchalski-Katzir, E. et al. Proc. Natl. Acad. Sci. USA 89 1992
Soft docking: matching of molecular surface cubes Jiang, F.; Kim, S J. Mol. Biol. 219(1) 1991
Use of GRID Potentials to Simplify the Docking¶
Instead of using explicit energy calculations, it is possible to use grid potentials, which substantially speeds up the process. For each atom type present in the ligand, a grid of energies is pre-computed. The potential at a given point is then calculated by interpolation from the surrounding grid points. With this procedure, the interaction energy is easily and rapidly calculated. This method is also suited for "virtual reality" simulations where the molecule is manually moved by the user on the screen while computing the corresponding interaction energy in real time.

Assessing Shape Complementarity Using Grid¶
In the Grid representation, shape complementarity can be computed as a number of overlapping surface grid voxels between interacting molecules. The grid voxels usually correspond to three types: surface, interior and exterior voxels. Docking algorithms aim at maximizing overlapping surface voxels and minimizing interior voxels. Some of these algorithms are presented later in this chapter.

articles
Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.; Aflalo, C.; Vakser, I. Proc. Natl. Acad. Sci. USA 89 1992
Soft docking: matching of molecular surface cubes Jiang, F.; Kim, S. J. Mol. Biol. 219 1991
wikipedia
Scoring Methods¶
Need to Assess the Quality of Docked Complexes¶
Scoring methods aim at assessing the quality of docked complexes and guiding the docking algorithm. The development of accurate scoring methods is considered to be critical for good quality results. In the following section we go into the basis of scoring methods and methodologies in some detail.

A Good Understanding of the Binding¶
Molecular recognition is a complex and subtle process. The development of a scoring method that can discriminate between good and bad binders is not a trivial task. In the example of the two enantiomers shown below and docked to the RAR protein, one is active and the other one is inactive. Would you be able to recognize which is which?

articles
Enantiomer discrimination illustrated by high-resolution crystal structures of the human nuclear receptor hRAR Klaholz B.P. et al. PNAS 97(12) 2000
Important Questions¶
What determines binding between a ligand and its receptor? What structural features are necessary? Is the pose acceptable? What is the most parsimonious way to sort a list of compounds in terms of their binding capabilities? All these issues, which are often difficult to grasp and simulate on a computer, depend to a great extent on our understanding at the molecular level, of the forces and energies governing the binding process.

Molecular Determinants for Binding¶
The binding process that leads to the formation of a complex between a ligand and its receptor is controlled by several factors including: (1) the interaction energies between the two molecules; (2) the desolvation and solvation energies associated with the interacting molecules and (3) the entropic factors that occur upon binding.

articles
Structural basis of macromolecular recognition Wodak SJ, Janin J. Adv Protein Chem. 61 2002 10.1016/S0065-3233(02)61001-0
Interaction Forces and Binding Energies¶
The binding forces associated to a complex between two molecules are based on the interaction energies between them. They represent thousands of elementary interactions and can be divided into two types: favorable and non-favorable interactions. The final free energy of binding will depend on the overall balance of these two types of interactions.

wikipedia
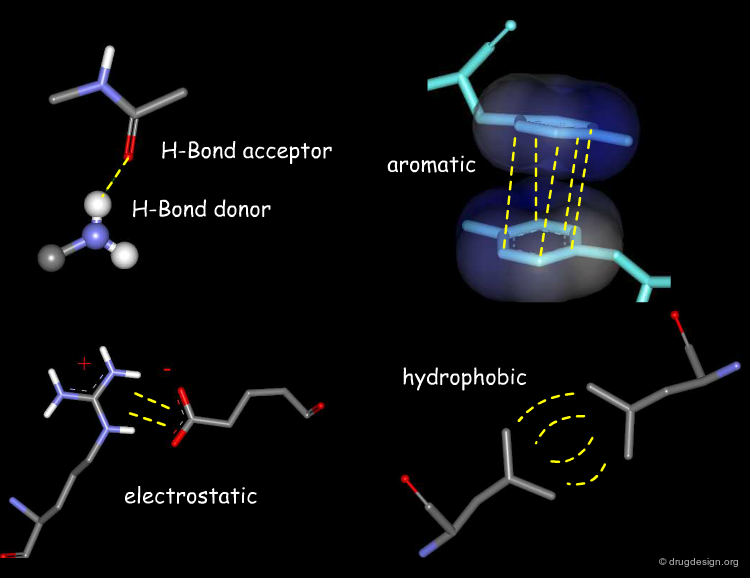
Favorable Forces¶
Favorable forces include good hydrophobic contacts, attractive electrostatic interactions and hydrogen bonds. The second and third types of interactions are strong and directional, whereas the first (favorable hydrophobic contacts) corresponds to locally weak and non-directional forces. However their accumulation on large surfaces may represent forces that can lead to substantial effects in terms of affinity and binding.

Unfavorable Forces¶
Unfavorable forces include bad hydrophobic contacts (steric clashes) and repulsive electrostatic interactions, which can all be assessed by energy calculations. A docking treatment aims at finding the relative orientation of the two molecules that corresponds to the lowest energy of interaction; therefore it aims at maximizing the favorable interactions and minimizes the unfavorable ones.

Desolvation Energies¶
The binding of a ligand to a protein is a complex process influenced by desolvation and solvation phenomena where the interacting entities become partially desolvated. This subtle thermodynamically driven chain of events leads to the formation of favorable interactions between the ligand and the protein where hydrophobic contacts are the driving forces: hydrophobic moieties associate together to reduce the interactions with the surrounding water.

wikipedia
Entropic Effects¶
The flexibility of the molecules and the consequences in terms of entropy can have a significant impact on the binding energy of a ligand where the solvent plays a crucial role by influencing the conformations of the interacting molecules. To account correctly for molecular flexibility the interaction of ensemble averages over many structures, and not only the interaction of two rigid entities has to be included.

wikipedia
Calculation of the Binding Energies¶
A complex has a lower potential energy than its constituent parts; this is what keeps them together. The binding energy ΔGbinding is the energy required to separate a complex into separate parts (protein and ligand); it is defined as the difference between the energy of the associated (bound) form (Ecomplex) and that of the separated (unbound) molecules (Eprotein and Eligand). Note that the energy of the complex (Ecomplex) alone has no significant meaning by itself. In the following pages we present methods for calculating ΔGbinding.

book
Reddy, M. Rami; Erion, Mark D. Free Energy Calculations in Rational Drug Design Springer - Verlag 2001
wikipedia
Free Energy Equations¶
The binding of a ligand to a protein is a reaction equilibrium with an equilibrium constant K that can be measured experimentally (equations 1 and 2). The relationship between K and ΔG, the free energy of binding, is obtained from thermodynamics (equation 3); whereas the reaction's free energy change ΔG is given by the Gibbs equation in terms of the variations of enthalpy ΔH and entropy ΔS (equation 4).

wikipedia
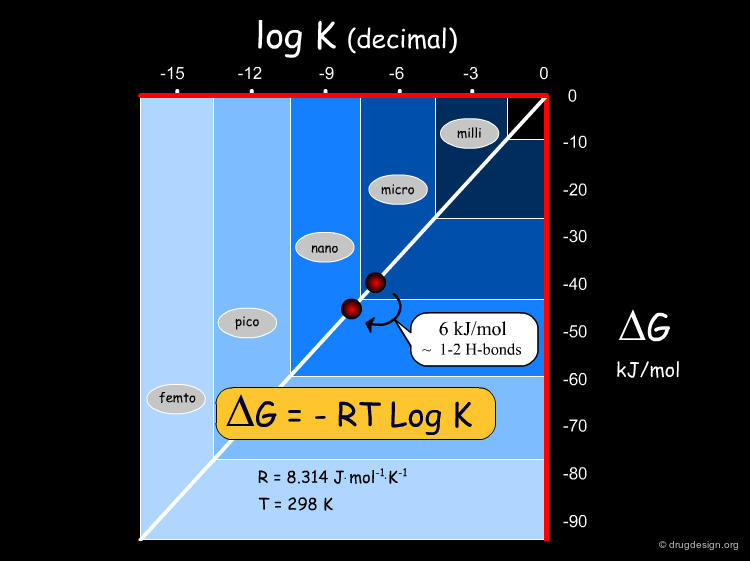
Conversion of K to Energies¶
Using the equation ΔG = -RT LogK, we can convert equilibrium constants into energies, and vice versa. The following diagram represents ΔG (in kJ/mol) as a function of log K. Note that a free energy of 6 kJ/mol corresponds to about one order in logK, which may represent 1-2 hydrogen bonds. This can make the difference between a micromolar and a nanomolar compound.
Difficulty of Calculating Free Energies of Binding ΔG¶
It is hard to estimate free energies of binding based on the Gibbs equation directly. This is mainly due to the difficulty of estimating the entropic component appearing in the second term of the equation. In a first approximation this entropic component is often neglected.

Approximating ΔG by Molecular Mechanics¶
By first neglecting the change in entropy and then using equation 2 where the pressure is considered to be constant (PΔV=0), the binding free energies ΔG can be approximated by the variation in the internal energy ΔU of the system. Molecular mechanics is a method that can estimate the internal energy of the system, which makes this approach practical for calculating ΔG. This is presented in the following pages.

wikipedia
Force-Field Calculations¶
Molecular mechanics is an approach based on the idea that the atoms of the molecule feel forces and the energy of a molecular system is related to these forces. An empirical set of energy functions called a force field can simulate such forces. The total energy of a system is described as the sum of the independent terms of the force field. Using the approximations indicated in the previous page, the energies obtained by force field methods can be used directly to approximate free energies of binding.

wikipedia
CHARMM Force Field to Score the Docking¶
The first version of the docking program AUTODOCK used the CHARMM force field to score the docking. It became very popular and extensively used because of the successful results obtained for many molecular dynamics simulations of proteins, membranes and nucleic acids that proved to be sufficiently accurate.

articles
CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S and Karplus M Journal of Computational Chemistry 4 1983 10.1002/jcc.540040211
Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function Morris, G. M., Goodsell, D. S., Halliday, R.S., Huey, R., Hart, W. E., Belew, R. K. and Olson, A. J. Computational Chemistry 19 1998 10.1002/(SICI)1096-987X(19981115)19:14%3C1639::AID-JCC10%3E3.0.CO;2-B
wikipedia
Approximating ΔG by Quantum Mechanics¶
Instead of using energies calculated by molecular mechanics, one idea was that they could be calculated by quantum mechanics. However, it rapidly became clear that the approach was too heavy computationally to produce easily exploitable results. Hybrid methods called QM/MM have developed that combine both quantum mechanics (QM) and molecular mechanics (MM) where critical aspects of the system are treated quantum mechanically and the bulk is treated classically by molecular mechanics, which results in acceptable computing times.

wikipedia
Development of Scoring Functions for Docking¶
The first force-fields such as CHARMM or AMBER that were used in molecular docking software were not developed for reproducing binding energies. In fact, they were used for molecular dynamics simulation of macromolecules. This prompted the idea of developing "scoring functions" that could be adapted to the needs of docking treatments. Based on a concept similar to the one used in force field simulations where the total energy of a system is described as the sum of independent terms, scoring functions were developed and parametrized in order to reproduce experimental binding energies.

articles
CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S and Karplus M Journal of Computational Chemistry 4 1983 10.1002/jcc.540040211
The Amber biomolecular simulation programs D.A. Case, T.E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K.M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods. J. Comput. Chem. 26 2005 10.1002/jcc.20290
Amber:assisted model building with energy refinement. a general program for modeling molecules and their interactions. P. K. Weiner and P. A. Kollman. J. Comput. Chem. 2 1981
Scoring Functions¶
Scoring functions calibrated for docking purposes can be classified into two major categories: (1) the empirical approach which is a force field method customized to docking and (2) the knowledge-based approach where interaction energies are calibrated for each type of atomic interaction. We discuss these in the following pages.

articles
Principles of docking: An overview of search algorithms and a guide to scoring functions Halperin I, Ma B, Wolfson H, Nussinov R. Proteins: Structure Function and Genetics Jun 1;47(4) 2002 10.1002/prot.10115
Molecular recognition and docking algorithms Brooijmans N, Kuntz ID. Annu Rev Biophys Biomol Struct 32 2003 10.1146/annurev.biophys.32.110601.142532
Structural basis of macromolecular recognition Wodak SJ, Janin J. Adv Protein Chem. 61 2002 10.1016/S0065-3233(02)61001-0
The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. Bohm HJ J. Comp. Aided Mol. Design 8 1994
Detailed Analysis of Scoring Functions for Virtual Screening Martin Stahl and Matthias Rarey J. Med. Chem. 44 2001
A Docking Score Function for Estimating Ligand-Protein Interactions: Application to Acetylcholinesterase Inhibition Jianxin Guo, Margaret M. Hurley, Jeffery B. Wright and Gerald H. Lushington J. Med. Chem. 47 2004 10.1021/jm049695v
Further development and validation of empirical scoring functions for structure-based binding affinity prediction Renxiao Wanga, Luhua Laib and Shaomeng Wanga Journal of Computer-Aided Molecular Design 16 2002
Consensus scoring for ligand/protein interactions Robert D. Clark, Alexander Strizhev, Joseph M. Leonard, James F. Blake, James B. Matthew Journal of Molecular Graphics and Modelling 20 2002 10.1016/S1093-3263(01)00125-5
Assessing Scoring Functions for Protein-Ligand Interactions Philippe Ferrara, Holger Gohlke, Daniel J. Price, Gerhard Klebe, and Charles L. Brooks J. Med. Chem. 47 2004 10.1021/jm030489h
Customized versus universal scoring functions: application to class I MHC-peptide binding free energy predictions A. Logean, A. Sette and D. Rognan Biiorganic and Medicinal Chemistry Letters 11 2001
Large-Scale Validation of a Quantum Mechanics Based Scoring Function: Predicting the Binding Affinity and the Binding Mode of a Diverse Set of Protein-Ligand Complexes Kaushik Raha and Kenneth M. Merz, Jr. J. Med. Chem. 48 2005
A Knowledge-Based Energy Function for Protein-Ligand, Protein-Protein, and Protein-DNA Complexes Chi Zhang, Song Liu, Qianqian Zhu, and Yaoqi Zhou J. Med. Chem. 48 2005
Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes Eldridge M and Auton TA J. Comp. Aided Mol. Design 11 1997
Empirical scoring functions. II. The testing of an empirical scoring function for the prediction of ligand-receptor binding affinities and the use of Bayesian regression to improve the quality of the model Murray, C. W.; Eldridge, M. D. J. Comp. Aided Mol. Design 12 1998
wikipedia
Empirical Scoring Functions¶
Empirical scoring functions are force fields calibrated to a particular training set of complexes with known 3D structures (X-ray data) and experimental biological activities. Each type of force field contribution is multiplied by a weighting coefficient (determined empirically by the parameterization study). In order to be computationally efficient some methods use a restricted number (subset) of force-field terms, for example "Ligscore" (derived from a training set of 122 complexes) uses only three terms: Van der Waals, polarity and desolvation.

articles
Further development and validation of empirical scoring functions for structure-based binding affinity prediction Renxiao Wanga, Luhua Laib and Shaomeng Wanga Journal of Computer-Aided Molecular Design 16 2002
Example of Empirical Scoring Function¶
AutoDock is an example of a program based on empirical scoring functions and was developed by Goodsell and Olson at the Scripps Clinic (California). The types of interaction energies included in AutoDock include hydrophobic contacts, electrostatic interactions, hydrogen bonds and solvent excluded volumes. More recently other terms were added to AutoDock to fit to a broader set of experimental data.

articles
Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function Morris, G. M., Goodsell, D. S., Halliday, R.S., Huey, R., Hart, W. E., Belew, R. K. and Olson, A. J. Computational Chemistry 19 1998
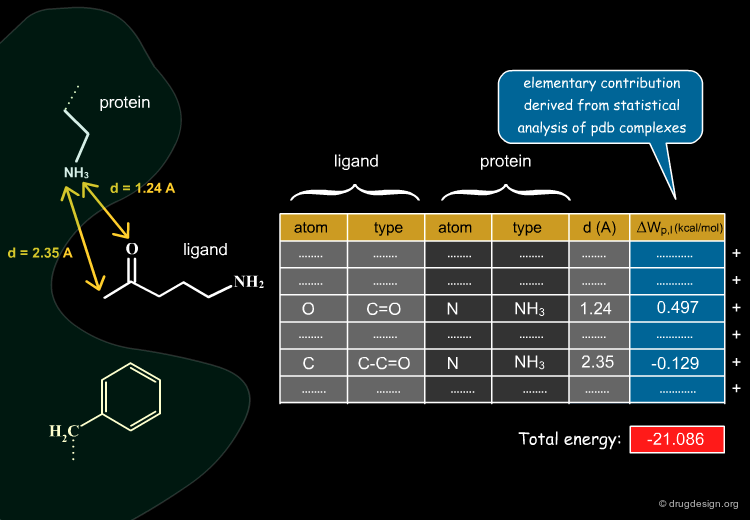
Knowledge-Based Scoring Functions¶
The knowledge-based approach is based on statistical analyses of X-Ray (PDB) complexes; its aims at deriving elementary energy contributions ΔWp,l for each type of non-bonded atom pair interaction between a protein (p) and a ligand (l). In the following pages we present how the ΔWp,l values are calculated.
articles
A general and fast scoring function for protein-ligand interactions: a simplified potential approach Muegge, I., Martin, Y.C. J. Med. Chem 42 1999
Predicting binding modes, binding affinities and 'hot spots' for protein-ligand complexes using a knowledge-based scoring function Gohlke, H., Hendlich, M., and Klebe G. Perspectives in Drug Discovery and Design 20 2000
Knowledgebased Scoring Function to Predict Protein-Ligand Interactions Gohlke, H., Hendlich, M., and Klebe, G. JMB 295 2000
Small Molecule Growth 2001 (SMoG2001): An Improved Knowledge- Based Scoring Function for Protein-Ligand Interactions Ishchenko, A.V. and Shakhnovich, E.J. J. Med. Chem. 45 2002
A Knowledge-Based Energy Function for Protein-Ligand, Protein-Protein, and Protein-DNA Complexes Chi Zhang, Song Liu, Qianqian Zhu, and Yaoqi Zhou J. Med. Chem. 48 2005
The Statistical Analyses¶
Statistical analyses calculate the distribution of occurrence frequencies in the distances for each type of pair of interactions (the more often an interatomic distance occurs, the more favorable it is). The figures bellow show the 3D distribution of protein-ligand contacts collected around reference amino acids for Arginine and Tyrosine.


Knowledge-Based Potentials¶
Distribution of occurrence frequencies can be translated into free energy scores for a given pair of atoms at a given distance using Boltzmann distributions. The interaction potential generated is usualy called "potential of mean force" (PMF) and is constructed for each type of interaction. For example in PMFScore 126 protein atom types and 34 ligand atom types are defined which give rise to 4284 potential curves.

wikipedia
The DrugScore Program¶
DrugScore is an example of a docking program using a knowledge-based scoring function. It was developed by G. Klebe at Philipps University in Marburg (Germany) that converted structural information contained in a set of about 1400 protein-ligand complexes taken from the Protein Data Bank. It has been used in many applications and proved to be able to successfully discriminate between incorrect models and crystallographically determined complexes.

articles
Statistical potentials and scoring functions applied to protein-ligand binding Golhe H and Klebe G. Curr. Opin. Struct. Biol. 11 2001 10.1016/S0959-440X(00)00195-0
Knowledge-based scoring function to predict protein-ligand interactions Gohlke H, Hendlich M, Klebe G J. Mol. Biol. 295 (2) 2000
DrugScore: The Thrombin Example¶
The picture below visualizes the per-atom DrugScore contributions of thrombin (deep blue, coated by the semi-transparent molecular surface) bound to an in-silico generated putative binding geometry of one inhibitor (melagatran, in green) and created with PyMOL. Favorably interacting atoms are surrounded by blue spheres whereas unfavorable interactions are shown in red. The sizes of the spheres correspond to the values of the contributing per-atom scores (a large blue sphere denotes a more attractive interaction than a small one; a large red sphere stands for more repulsive energy than a small one).

Refinement of Scoring Functions¶
Scoring functions are sensitive to the proteins represented in their training sets and are unable to treat all types of proteins equally well. Scoring function development is a continually expanding field aiming at better assessing both poses and energies. Empirical scoring functions can be refined, the same way that force fields were refined; however, knowledge-based scoring functions that rely on statistical analyses of a huge set of experimental complexes cannot be easily refined in a stepwise manner.

articles
Novel scoring methods in virtual ligand screening. Pick D. Methods Mol Biol. 275 2004
book
Pick D. Chemoinformatics: Concepts, Methods and Tools for Drug Discovery Humana Press 2004
Other Scoring Methods¶
Many other methods can also be used to estimate protein binding affinities. A diagram of the existing methods is shown below ranked in terms of their quality and applicability in high throughput treatments. The most accurate approaches are usually more demanding in terms of computational cost. Some of them are briefly presented here.

Shape and Property Complementarity Scoring¶
Many docking algorithms use shape complementarity as their primary score. Other matching properties can be combined to shape complementarity and include physico-chemical descriptors such as electrostatic energy, H-bonds, solvation, hydrophobicity, pharmacophores matching, etc... Contrary to energy-based calculations these methods focus on ligand-receptor complementarity and are therefore suited to measure the goodness of fit or to rank solutions, rather than estimating absolute binding free energies.
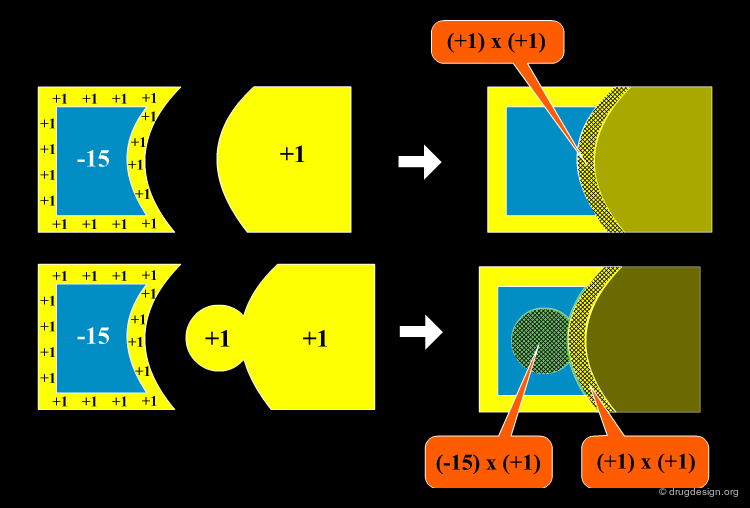
Method to Measure Shape Complementarity¶
A number of methods have been developed to quantify the shape complementarity between molecules. The simplest one is illustrated here. The molecules are introduced into a 3D grid. Negative scores (in blue) are given to the interior of the first protein, and positive scores (in yellow) are given to the border of both proteins and the interior of the second one. The shape complementarity score is the sum of the products of all cells. Other, and more sophisticated methods include the FFT approach (presented later) and the statistic Sc method (Lawrence and Colman, 1993).


Free Energy Perturbation¶
Free Energy Perturbation is the most rigorous method for calculating binding energies. It takes advantage of the properties of thermodynamic cycles and aims to predict the free energies of binding of new ligands from knowledge of the experimental binding energy of a known reference compound. The method cannot be routinely used for docking applications for the following two reasons: (1) it requires substantial computational time even for one single ligand; (2) it gives good results principally when the structural changes between the molecules to be compared are relatively small. FEP is presented in more detail in the chapter on molecular dynamics.


book
Reddy, M. Rami; Erion, Mark D. Free Energy Calculations in Rational Drug Design Springer - Verlag 2001
wikipedia
Rigid Docking Methods¶
Docking Algorithms¶
We now discuss the core features of docking algorithms. In this section we present docking algorithms which consider molecules to be rigid entities. Docking methods taking flexibility into consideration are presented in the next section.

The Mathematical Problem¶
The mathematical problem can be defined as follows: if we assume that the molecules are rigid, we are then looking for a transformation in 3D space of one of the molecules which brings it into optimal fit with the other molecule in terms of a scoring function. The search space is restricted to three rotational and three translational degrees of freedom.

Two Docking Philosophies¶
A large number of docking approaches have been developed to predict the formation of molecular complexes. They can be divided into two broad classes that correspond to two different philosophies: feature-based matching and the stepwise search. The first approach is derived from computer vision and matches local complementarity features among molecules involved in the recognition. The second class of approaches explores the 'search space' guided by a scoring function.

The Feature-Based Matching Approach¶
"Feature-Based Matching" is an approach derived from computer vision for object matching. It tries to recognize similarity features between a "model image" (in our case a ligand) and the "scene" (a protein). It is used in many areas including image analysis, robotics, pattern recognition and computational docking.

Docking Using Feature-Based Methods¶
The process of docking two rigid molecules can be described by matching characteristic ligand features to complementary protein features in the molecular space. "Features" can consist of volume segments of the ligand or the protein or complementary interactions such as hydrogen bond donor and acceptor. Feature-based matching methods are based on algorithms that are pathway independent.

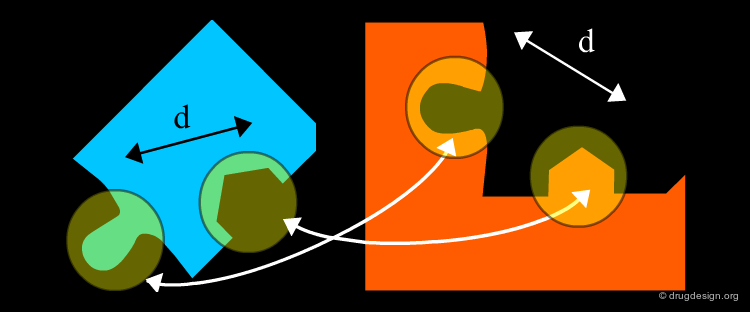
Match Complementarity or Similarity Features¶
In the feature-based matching approach the match can be based either on complementarity features or on similarity features. For example when trying to match the shapes of the two molecular surfaces, the matching function should search for complementarity features (knob and hole, concave and convex, etc...). Alternatively if we are trying to match pharmacophoric elements derived from the receptor, the matching function should search for similar features.

Components of Feature-Based Matching Methods¶
The way feature-based matching applies to the docking problem is similar to solving a jigsaw puzzle. You pick a piece and look for a complementary one from among the rest of the pieces (feature extraction). Once a piece is found (feature matching), the elements are assembled (transformation), the solution is then assessed globally for final approval of the compatibility (filtering and scoring).

Step 1: Feature Extraction¶
In the first step, features of interest are extracted or generated from the molecular structures of both molecules independently. Example of features are surface descriptors, volume, points and distances, bond vectors, grid and various properties such as electrostatic potential, hydrophobic moment, polarity, atom-types, H-bond donor, H-bond acceptor etc. It is crucial to identify which features are relevant to the problem at hand.

Step 2: Feature Matching¶
In step two, a preliminary list of candidate pairs of corresponding features is drawn up. Then using a matching algorithm, a list of feature pairs consistent with an object model is calculated. Consistency is generally assessed in terms of distances between features of both molecules.
Step 3: Transformation (Assembly)¶
The third step consists of applying the geometrical transformation defined by the solution found in the previous step and resulting in the assembly of the two elements. Note that some methods start with transformation generation and derive the feature match list.
![]()
Step 4: Filtering and Scoring¶
The last step is filtering and scoring. Filtering aims at removing solutions that contain unacceptable matches; since the transformations were computed by considering only local features they may include steric clashes or other incompatibilities. The resulting matching needs to be assessed globally and then ranked.

Virtual Screening and De Novo Design¶
Feature matching methods can be used to scan a library of molecules. The approach is named "de-novo design" when the library contains fragments of molecules. It is termed "virtual screening" when the library contains real molecules. In general virtual screening is straightforward: the hits can be readily ordered and tested. De-novo design is more creative, but requires the synthesis of new molecules; it will be presented in another chapter of this course.


wikipedia
Programs with Feature-Based Matching Methods¶
The pioneer DOCK program (Kuntz, 1982) was developed with a feature-based matching approach. A large number of docking programs are based on this principle. Some examples of docking programs using the feature-based matching approach are listed in the table below.

articles
A geometric approach to macromolecule-ligand interactions Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 161 1982
Algorithms of Matching¶
Beyond the choice of features to be compared, one of the key components of a feature-based program consists of its matching algorithm whose role is to identify potential matches between objects, based on sets of points associated with properties (see picture below). To apply feature-based matching methods in virtual screening, the feature matching algorithm must be very efficient. The clique and pose-clustering matching algorithms are presented in the following pages.

Clique-Search Based Approaches¶
DOCK, the pioneering docking program developed by Kuntz et al. reduces the docking problem to matching the ligand atom points to the negative image of the receptor, based on distances between the points. The shape of the receptor pocket is described by spheres, and the centers of the spheres are regarded as potential locations for ligand atoms. At least four ligand atoms must match individual sphere centers to count as a valid ligand match.



articles
A geometric approach to macromolecule-ligand interactions Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 161 1982
Goal of the Docking Algorithm¶
The aim of the DOCK algorithm is to find a transformation in 3D space that maximizes the fit of the ligand with respect to the receptor. A simple but rather time-consuming approach consists of trying every possible matching. This cannot be used routinely with today's large databases. A more efficient alternative is to use a "docking graph" and search for maximum clique on this graph. This is explained in the following page.
articles
A geometric approach to macromolecule-ligand interactions Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 161 1982
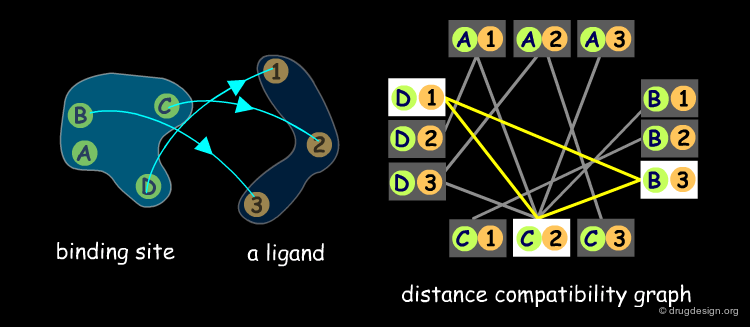
Distance Compatibility Graph¶
The matching procedure used in the DOCK program exploits the clique detection algorithms developed by Bron and Kerbosch (1973). A "clique" is a concept derived from graph theory. A distance compatibility graph is constructed where each node corresponds to pairing receptor and ligand points. Each edge corresponds to a match of size 2, with equal distances between site points. For example in the following graph B-1 and C-2 are compatible (they could be superimposed) and are visualized in the graph by an orange line.

articles
A geometric approach to macromolecule-ligand interactions Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 161 1982
Algorithm 457: Finding all cliques of an undirected graph. Bron C., Kerbosch J. Communications of the ACM 16 1973
Clique Detection Methods¶
When the distance compatibility graph is constructed, the clique detection method searches a fully connected sub-graph (also called a clique). In the example below there is only one clique (shown in yellow), the three nodes D1, C2, B3, are connected together, and represent a solution to our matching problem.
articles
A geometric approach to macromolecule-ligand interactions Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 161 1982
Algorithm 457: Finding all cliques of an undirected graph. Bron C., Kerbosch J. Communications of the ACM 16 1973
Pose-Clustering¶
Pose clustering is another algorithm for matching structural features. The algorithm tries to match a triplet of features from the first object (receptor) with a triplet of features from the second one (ligand). This approach is similar to the one used in computer vision for matching local features.

articles
J. of Computer Vision, Graphics, and Image Processing Stockman, G.
40 1987
Molecular surface recognition by a computer vision-based technique Norel, R.; Fischer, D.; Wolfson, H.; Nussinov, R Protein Eng 7 1994
book
Rarey, M.; Kramer, B.; T., Lengauer 3rd Int. Conf. on Intelligent Systems for Molecular Biology (ISMB'95) AAAI Press 1995
Searching for Compatible Triangles¶
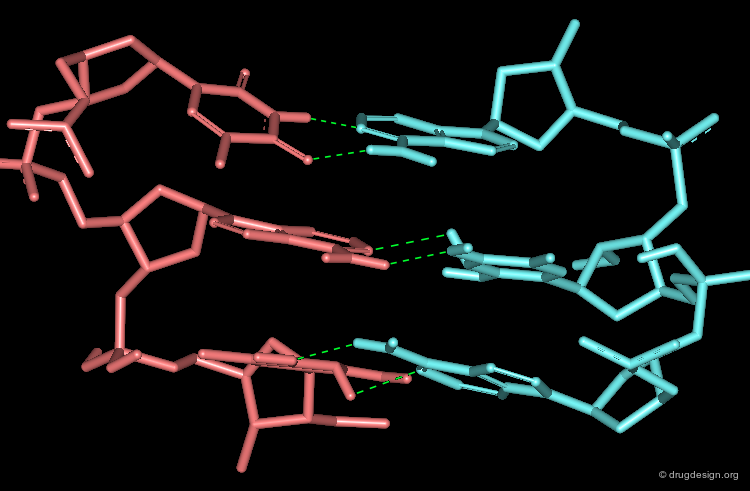
A triangle pair is necessary in order to define a transformation. The first step in the pose-clustering algorithm is to define all possible triangles of functional groups of the ligand and functional groups of the protein. Each triangle of the receptor is then matched with all those of the ligand. Matching features must satisfy two conditions: (1) functional group interactions must be compatible (e.g. charges or H-bonds donor/acceptor); (2) the triangles must be approximately of the same geometry. For each matching triangle the transformation is calculated and stored.

Transformation that Align a Maximum of Triangles¶
The process of triangle matching generates many transformations, some of which are similar. A clustering step clusters all similar transformations and ranks clusters with a high number of matching triangles. Large clusters indicate a region with a high number of matching features: they correspond to potential docking solutions.

wikipedia
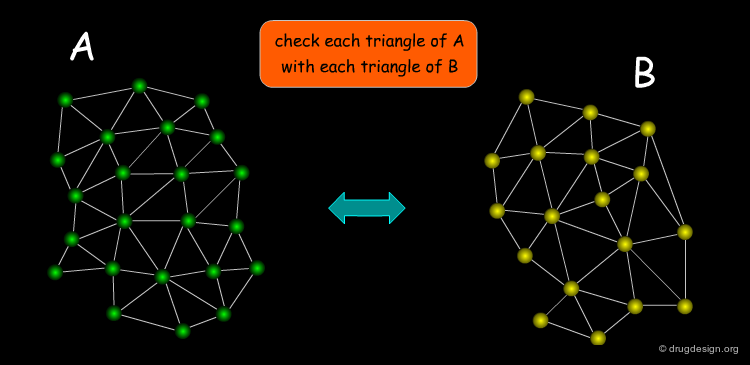
Complementarity and Similarity Matching¶
As previously mentioned, the feature-based matching method can be based either on complementarity or similarity features. An illustration of complementary and similarity matches in pose clustering is shown here. The Delaunay triangulation method has been used to transform the protein surface into sets of triangles.


wikipedia
Speed up of Pose-Clustering¶
Note that it is possible to speed up the pose clustering by using two points and their normals (instead of three points) in order to compute the transformation; this considerably reduces the computing time. The complexity of the two methods are indicated in the following view; n and m are the number of features in the receptor and the ligand respectively.

articles
Molecular surface complementarity at protein-protein interfaces: the critical role played by surface normals at well placed, sparse, points in docking Norel, R.; Lin, S.; Wolfson, H.; Nussinov, R J. Mol. Biol 252 1995
The Bottleneck of Pose-Clustering¶
One of the bottlenecks of the pose-clustering approach is the inefficiency of the matching search step where all triangles of the receptor are tested for matching with all possible triangles of the ligand. This issue makes the high throughput docking of a library of ligands using a pose-clustering approach inefficient. Geometrical hashing is a method that can be used to improve this problem.
Geometric Hashing¶
An efficient technique for object recognition named "Geometric Hashing" was introduced in computer vision by Lamdan et al. The idea consists of using a database where the features of known geometric objects are stored, which enables the rapid recognition of a query object. This technique has been applied with great success in a variety of areas including medical imaging, computational molecular biology and computer-aided drug design.

articles
A geometry-based suite of molecular docking processes Fischer, D.; Lin, S.; Wolfson, H.; Nussinov, R J. Mol. Biol 248 1995
book
Lamdan, Y.; Wolfson, H. Proceedings of the IEEE Int. Conf. on Computer Vision
1988
Wolfson, H. Proceedings of the European Conf. on Computer Vision
1990
wikipedia
# Fast Retrieval of Matching Features¶
Its use in computational docking was first introduced by Nussinov and Wolfson in 1991 for fast retrieval of matching features. In a preliminary phase the geometrical features of the ligand are stored with an efficient indexing-based method (hash tables). Then the treatment searches for all matches by using the hash tables. The example below illustrates the fetching of triangles ADD (Acceptor, Donor, Donor).

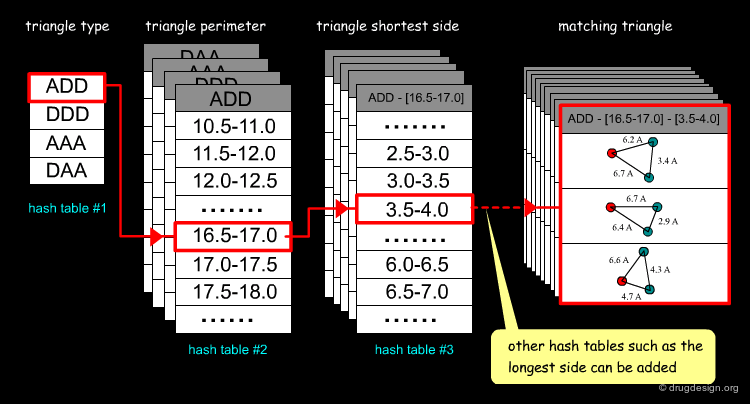
book
Volker Schnecke and Leslie A. Kuhn RIGIDITY IN THEORY AND APPLICATIONS PLENUM PUBLISHING, 1999
wikipedia
Media
This picture was adapted from the article of Volker Schnecke and Leslie A. Kuhn "FLEXIBLY SCREENING FOR MOLECULES INTERACTING WITH PROTEIN" see reference
# Invariant Representation of Features¶
One important issue in feature recognition is the dependence on the orientation of the object. To obtain an invariant representation with respect to the global position and orientation of a structure, geometric hashing method stores information invariant to rotations and translations. This is done for each triangle: a reference frame is defined, the coordinates of all the other points in this reference frame are calculated and then stored in the hash table.

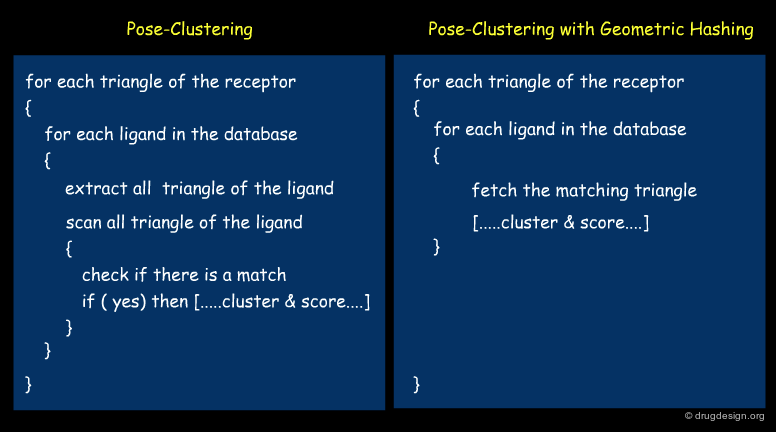
Improvement of Pose-Clustering¶
Geometric hashing is a method that can be used to speed up pose clustering. Each molecule of the library is preprocessed and transformed in a hash table so that, based on a specific query, it is easy to find all triangles of geometry X, property Y in the molecule Z. The preparation of the hash table is time consuming but it only needs to be done once. For each ligand all transformations are stored and clustered for matching features (using triangles or point pairs and normals).
articles
Flexible docking allowing induced fit in proteins: insights from an open to closed conformational isomers Sandak, B.; Wolfson, H.; Nussinov, R Proteins: Structure Function and Genetics 32 1998
book
Lenhof, H Proc. of the First Annual International Conference on Computational Molecular Biology
1997
Duhovny, D.; Nussinov, R.; Wolfson, H Workshop on Algorithms in Bioinformatics Springer Verlag: 2002
PatchDock Example¶
PatchDock is an example of software that combines pose-clustering matching and geometric-hashing techniques for protein-protein docking. First the PatchDock algorithm divides the Connolly dot surface representation of the molecules into concave, convex and flat patches. Then, complementary patches are matched in order to generate candidate transformations. Each candidate transformation is further evaluated by a scoring function that considers both geometric fit and atomic desolvation. Finally, an RMSD (root mean square deviation) clustering is applied to the candidate solutions to discard redundant solutions.
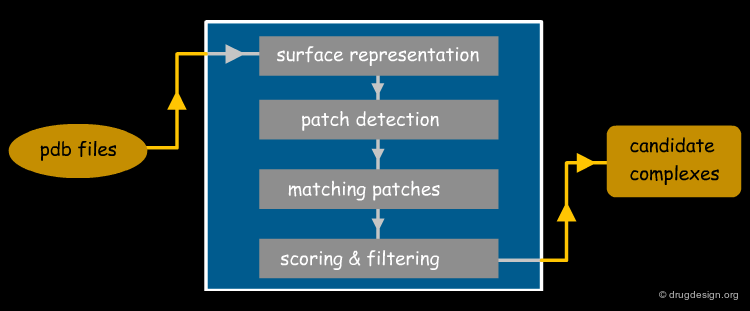

articles
PatchDock and SymmDock: servers for rigid and symmetric docking Schneidman-Duhovny D. et al. Nucleic Acids Research 33 2005 doi:10.1093/nar/gki481
The Stepwise Search Approach¶
The stepwise search approach tries to explore the 'search space' (defined as the set of all possible solutions), with the hope of finding an optimal solution. The approach is driven by a scoring function which guides the search algorithm.
Components of a Stepwise Docking Program¶
In computational docking the stepwise search involves two components: (1) a "positioning module" which generates new complex arrangements and (2) a "scoring module" that assesses the quality of each individual arrangement. The positioning module is directly connected to the search module, which dictates the configuration of the next pose to be generated. In rigid docking the variables to optimize are the three rotation angles and the three translation parameters.

Exhaustive and Stochastic Search¶
The search algorithm can be either exhaustive (also called 'brute force search'), where all possible docking configurations are systematically generated, or partial, where only a small part of the space is sampled randomly. Approaches based on random trials are called stochastic methods.

Exhaustive vs. Stochastic Search¶
An important difference between exhaustive and stochastic methods is that an exhaustive search will always find the optimal solution, whereas stochastic algorithms cannot guarantee they will find the best solution. Stochastic methods are said to be heuristic, which means that they are based on trial and error explorations.

wikipedia
Exhaustive Search¶
Exhaustive search represents the simplest and most intuitive search method. The receptor molecule usually remains stationary. The entire 6-dimensional space of ligand transformations is explored in order to find orientations that maximize surface complementarity. Exhaustive methods represent a formidable computational challenge and they cannot be applied if the search space is extremely large. In such cases stochastic methods are used.

articles
Soft docking: matching of molecular surface cubes Jiang, F.; Kim, S J. Mol. Biol. 219(1) 1991
BiGGER: a new (soft) docking algorithm for predicting protein interactions Palma, P.N., Krippahl, L., Wampler, J.E., Moura, J.J. Proteins: Structure Function and Genetics 39 2000
PUZZLE: a new method for automated protein dockingbased on surface shape complementarity Helmer-Citterich M. and Tramontano A. J.Mol.Biol 235 1997
ESCHER: a new docking procedure applied to the reconstruction of protein tertiary structure 1. Ausiello,G., Cesareni,G. and Helmer-Citterich,M Proteins: Structure Function and Genetics 28 1997
EUDOC: a computer program for identification of drug interaction sites in macromolecules and drug leads from chemical databases Pang, Y.; Perola, E.; Xu, K.; Prendergast, F. J. Comput. Chem 22 2001
Mapped-Grid Method¶
A grid approach can be used for exhaustive searches where the volumes of the receptor and ligand are mapped into a grid consisting of a three dimensional lattice of regularly spaced points. The receptor is fixed in this lattice, which makes it possible to sample the space with a finite number of points and to pre-calculate (and store) many physico-chemical properties of the receptor that are ready to be used in the simulation phase.

Physico-Chemical Properties of the Receptor¶
The calculation of the physico-chemical properties of the receptor is usually done with a probing technique, where a probe is placed at each point on the grid and is used to quantitatively measure the value of a field (van der Waals, electrostatic, etc...) created by the receptor at the point in question. This information is calculated once; it is used afterwards to assess the interaction with the ligand.

Assessing Shape Complementarity¶
The ligand is placed into the grid, with an orientation (rotation and translation) that varies at each step. The shape complementarity between the molecules is evaluated by counting the number of overlapping surface voxels between the two molecules. Overlapping interior voxels (penetration/clashes) are penalized in the overall count enabling the elimination of many unfeasible transformations.

wikipedia
Fast-Fourier Transform (FFT) Method¶
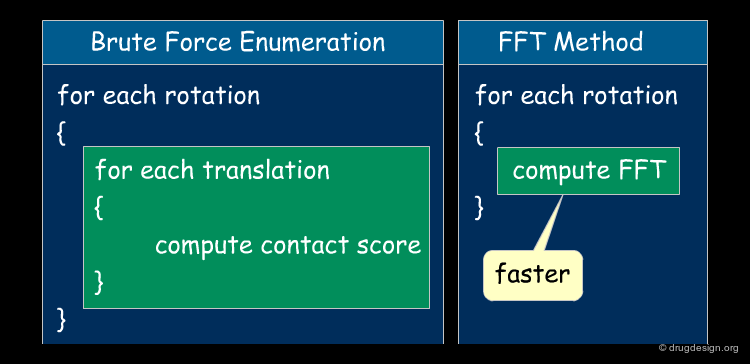
Katchalski-Katzir et al. introduced the FFT approach to brute force enumeration in 1992. This approach provides an efficient way of systematically exploring the space of docked conformations by speeding up the search in the 6-dimensional transformation space.

articles
Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA Proc Natl Acad Sci USA 89 1992
FFT vs. Exhaustive Method¶
In FFT-based approaches the rotational parameter space is scanned as previously by a brute force search. The FFT is used for fast enumeration of the translations. This means that instead of scanning all possible translations (Tx,Ty,Tz), the FFT is computed. As a result, the calculation of the contact scores for all possible orientations ends up on the order of O(n3logn), instead of O(n6), where n is the number of grid voxels. Note that the calculation of the contact score is integrated in the FFT module.
wikipedia
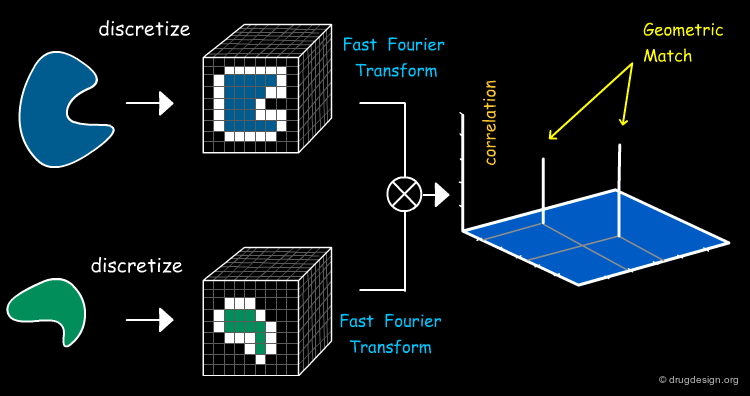
FFT - Geometric Shape Complementarity¶
In FFT-based approaches the geometric complementarity score is computed by a Fourier correlation. A high correlation score denotes good surface complementarity between the molecules.
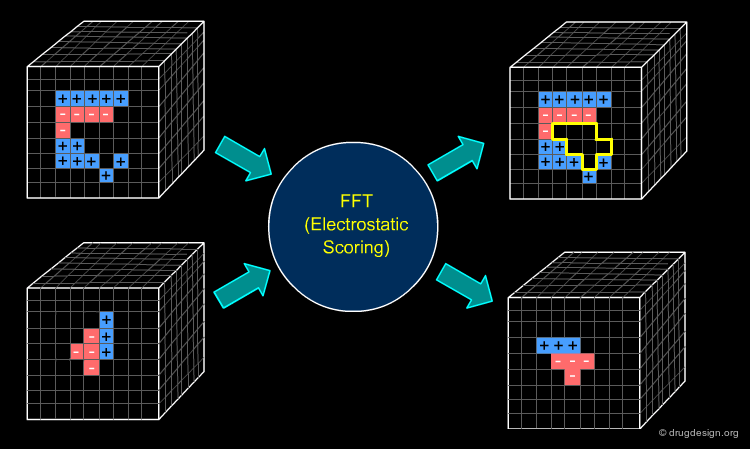
FFT - Different Scores¶
Many docking algorithms employ an FFT-based approach for fast evaluation of geometric shape complementarity. The electrostatic interaction can also be measured by a Fourier correlation. The simplest approach constructs the grids of the receptor and the ligand as follows. The electric field is computed at each grid voxel of the receptor molecule. The charges are assigned to the atoms of the ligand molecule and then discretized in a grid. A similar treatment is applied to Van der Waals interactions. Note that other scores, like desolvation, can be computed by FFT as well.
articles
Rapid refinement of protein interfaces incorporating solvation: application to the docking problem Jackson RM, Gabb HA, Sternberg MJE: J Mol Biol 276 1998
New algorithm to model protein-protein recognition based on surface complementarity : Applications to antibody-antigen docking Walls, P.; Sternberg, M J. Mol. Biol 228 1992
A systematic study of low-resolution recognition in protein-protein complexes Vakser, I.; Matar, O.; Lam, C. Proc. Natl. Acad. Sci. USA 96 1999
Protein docking using continuum electrostatics and geometric fit Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, Ten Eyck LF Protein Eng 14 2001
Docking unbound proteins using shape complementarity, desolvation, and electrostatics Chen, R.; Weng, Z Proteins: Structure Function and Genetics 47 2002
Electrostatics in protein-protein docking Heifetz, A.; Katchalski-Katzir, E.; Eisenstein, M Protein Sci 11 2002
Docking of Plastocyanin and Cytochrome C¶
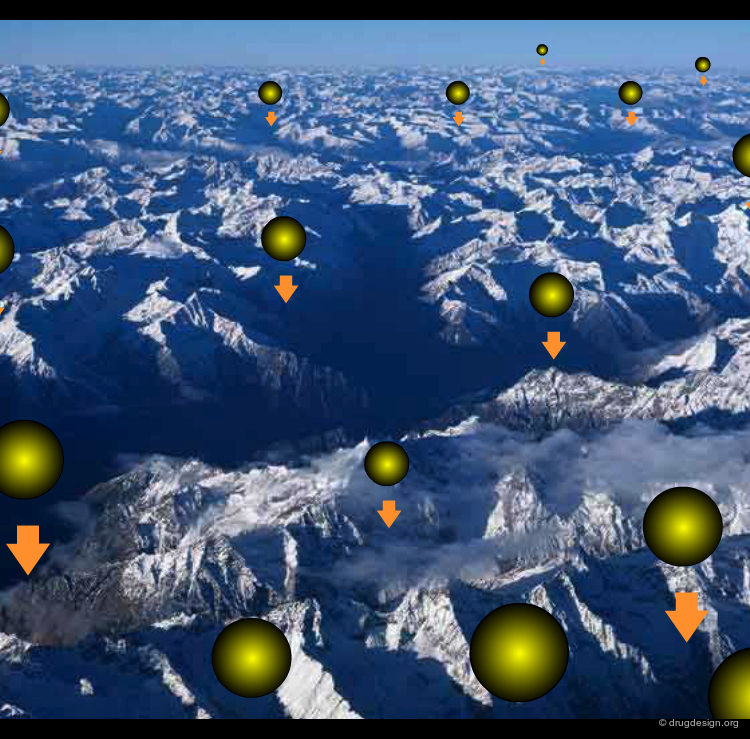
An example of FFT-based docking between plastocyanin and cytochrome-C, using the DOT software is shown here. The purpose of DOT is the rapid computation of the electrostatic potential energy between two proteins. It took only eight minutes of computing time using a cluster of 256 Intel computers to evaluate 2.6 billion different configurations. The spheres shown in the "Docking Results" button represent the centers of the 2,000 most favorable configurations found by DOT for cytochrome-C.


Spherical Polar Fourier Correlations - Fast FFT¶
Recent improvements by Ritchie et al. use spherical polar Fourier correlations to make the transformational search even faster. Unlike other FFT-based approaches, there is no need to calculate a new FFT for each possible rotation, since spherical harmonic functions transform under rotations. The six-dimensional transformation space is divided into five rotational angles and one distance. Both receptor and ligand are rotated. The receptor is translated along the z-axis. This approach is much faster than the original FFT, and electrostatics correlations may be calculated as well.

articles
Protein docking using spherical polar Fourier correlations Ritchie, D.; Kemp, G Proteins: Structure, Function and Genetics 39 2000
wikipedia
Stochastic Algorithms¶
Stochastic methods are suitable for solving complex optimization problems with many variables. These methods are well suited for finding the global optimum. They are based on the idea that a good sampling of the molecular space may be sufficient to converge towards an acceptable solution. Applied with success in many areas (business, economy, engineering, science), stochastic approaches are now extensively used in docking programs.
articles
Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations Gray, J.; Moughon, S.; Schueler-Furmn, O.; Kuhlman, B.; Rohl, C.; Baker, D J. Mol. Biol 331 2003
HADDOCK: A protein-protein docking approach based on biochemical or biophysical information Dominguez, C.; Boelens, R.; Bonvin, A. J. Amer. Chem. Soc 125 2003
Development and validation of a genetic algorithm for flexible docking Jones, G.; Willet, P.; Glen, R.; Leach, A J. Mol. Biol 267 1997
A Typical Computational Docking Program¶
A typical docking program generates a starting pose, and then small modifications (also called a "move") are made in an attempt to find an improved solution. If the resulting pose has a better score, then another step is made in the same direction, otherwise the new orientation is rejected and other candidate poses are generated and assessed using the same procedure. The process is continued until no improvement in the score can be obtained. The best score is assumed to represent the global minimum of the complex between the two molecules.

Optimization Methods to Find the Best Solution¶
Several stochastic approaches have emerged as efficient tools for solving complex optimization problems and have been extensively used in computational docking. They include methods such as Monte Carlo, Genetic Algorithms, Simulated Annealing and Tabu searches. We present these methods in the following pages.

Monte Carlo Methods¶
The Monte Carlo (MC) method provides approximate solutions to a variety of mathematical problems by performing statistical sampling experiments. In each step a random move is applied to the system, following a selection procedure that accepts or rejects the move. The chance of finding the best configuration increases with the number of steps that are made in the simulation. Many docking methods include some MC based sampling.

articles
ICM: a new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation Abagyan, R.; Totrov, M.; Kuznetsov, D. J. Comput. Chem. 15(5) 1994
J. Mol. Biol Gray, J.; Moughon, S.; Schueler-Furmn, O.; Kuhlman, B.; Rohl, C.; Baker, D.
331 2003
Software package for protein modeling and docking Trosset, J.; Scheraga, H. J. Comput. Chem. 20 1999
Distributed Automated Docking of Flexible Ligands to Proteins: Parallel Applications of AutoDock 2.4. Morris, G. M.; Goodsell, D.; Huey, R.; Olson J. Computer-Aided Molecular Design 10 1996
A Monte Carlo simulation approach to the molecular docking problem Liu, M.; Wang, S J. Computer-Aided Molecular Design 13 1999
MOLECULAR RECOGNITION AND DOCKING ALGORITHMS Natasja Brooijmans, Irwin D. Kuntz Annual Review of Biophysics and Biomolecular Structure 32 2003 10.1146/annurev.biophys.32.110601.142532
wikipedia
Simulated Annealing¶
Simulated annealing is a technique derived from Monte Carlo methods where the system is heated and then cooled very slowly. Heating allows the system to cross energy barriers and visit the whole conformational surface. Cooling drives the system into an energy minimum. This process is generally repeated many times until several very closely related and low energy solutions are obtained. The configuration of the lowest energy is assumed to be the exact docked solution. AutoDock was the first docking program to implement the simulated annealing approach.

Genetic Algorithms (GA)¶
Genetic algorithms (GA) are search techniques introduced in the 1960s by Holland to solve complex optimization problems based on evolutionary principles of natural selection and genetics. The underlying principle is the belief that the new population will be better than the old one. A genetic algorithm was first implemented in a computer program in 1992, since then it has been exploited in many applications. GA are well suited for parallel computing.
Voir Gold

articles
GAPDOCK: A genetic algorithm approach to protein docking in CAPRI round 1 Gardiner, E.; Willett, P.; Artymiuk, P Proteins: Structure, Function and Genetics 44 2001
Development and validation of a genetic algorithm for flexible docking ] Jones, G.; Willet, P.; Glen, R.; Leach J. Mol. Biol 267 1997
Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation Jones, G.; Willet, P.; Glen, R J. Mol. Biol 245 1995
Automated docking using Lamarckian genetic algorithm and an empirical binding free energy function Morris, G.; Goodsell, D.; Halliday, R.; Huey, R.; Hart, W.; Belew, R.; Olson J. Comput. Chem 19 1998
MOLECULAR RECOGNITION AND DOCKING ALGORITHMS Natasja Brooijmans, Irwin D. Kuntz Annual Review of Biophysics and Biomolecular Structure 32 2003 10.1146/annurev.biophys.32.110601.142532
book
Holland, J. H.
University of Michigan Press. 1975
wikipedia
General Principle of GA¶
The genetic algorithm begins with an initial set of populations that encodes some potential solutions (not optimal). At each step "genetic operators" are used to generate a new and better population by simulating in-silico the evolution process. After enough iterations the process is expected to converge to an optimal solution.

Creating a New Generation¶
To be included in a new generation two cases are possible: (1) natural selection dictated by the scoring function (also known as "fitness function") or (2) by being the outcome of the reproduction of the two parents. Those with the highest score are selected to be the parents for the next generation.

wikipedia
Simulating the Reproduction Process¶
The two genetic operators used to simulate the reproduction process are crossover and mutation. During reproduction, genes from parents combine (or crossover) to form a whole new chromosome. The newly created combination can then be mutated. Mutation means that the elements of DNA are changed slightly. These changes are mainly caused by errors in copying genes from parents. Chromosomes are represented here by binary strings encoding the rotation and translation variables to optimize.

Steps in Genetic Algorithms¶
A typical genetic algorithm involves the following steps: a random initial population encoding solutions to the problem is created. Genetic operators (e.g. crossover, mutation), are then applied to this population leading to a new population. This new population is then scored and ranked; their probabilities of getting to the next iteration round depend on their actual score. The process is repeated until either a maximum number of generations have been produced, or a satisfactory fitness level has been reached for the population.


Lamarckian Genetic Algorithm¶
Genetic algorithms are often combined with local searches to improve performance and convergence speed. Lamarckian Genetic Algorithm (LGA) is an example of such hybrid global-local optimization: each GA generation is followed by local adaptive search, which is performed on some part of the population. LGA is used by AutoDock method. The performance of LGA has been shown to be better than the performance of the previous AutoDock search method, Monte Carlo simulated annealing and traditional GA.

articles
Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function Morris, G. M., Goodsell, D. S., Halliday, R.S., Huey, R., Hart, W. E., Belew, R. K. and Olson, A. J. Computational Chemistry 19 1998
wikipedia
Tabu Search¶
Tabu search (TS) is a stochastic searching algorithm that maintains a list of previously visited poses. These poses are forbidden and cannot be revisited (they are "taboo"). TS effectively guides the search process into unvisited areas of the space and has been successfully implemented in a number of docking programs.

articles
Future paths for Integer Programming and Links to Artificial Intelligence Glover F. Computers and Operations Research 5 1986
Tabu Search: A Tutorial Glover F. Interfaces 20(4) 1990
book
Glover F. and D. de Werra
Baltzer Science Publishers 1993
Glover F., and Laguna M. Modern Heuristic Techniques for Combinatorial Problems John Wiley and Sons 1993
Glover, F. and M. Laguna
Kluwer Academic Publishers, Norwell, MA 1997
wikipedia
Tabu Algorithm¶
Tabu Search is based on neighborhood search, which means that at each step the algorithm considers the set of all possible neighborhood states and chooses the best one even though it might be worse than the current move. The best new solution (not in the Tabu list) is then chosen as the new current solution and the same process is repeated again. Not re-visiting previously visited states helps in two ways: (1) avoiding being trapped in local minima and (2) a better exploration of the space.
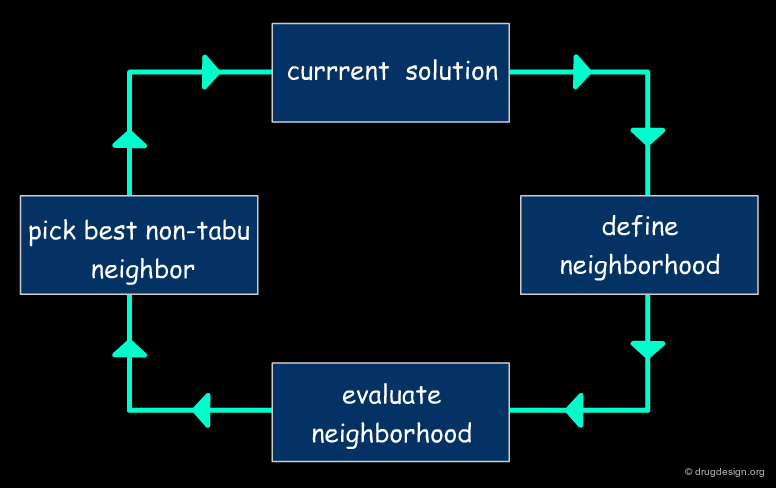
Avoiding Being Trapped in a Local Minimum¶
The main problem of methods based on local search approaches is that one can be trapped in a local minimum. By preventing the search from revisiting previously explored solutions the Tabu search avoids cycling over and over again into the same loop. But this not enough to escape this local minimum. To do so the Tabu search algorithm allows moves that deteriorate the current solution, with the hope of eventually identifying a better one.

Better Exploration of the Space¶
The second problem affecting methods based on local search approaches is that they tend to be biased in their search: they scan the space of solutions in a biased manner. The use of the Tabu algorithm can guide the search to ensure diversity in the exploration by increasing the chances of finding an optimal solution. To do so the Tabu search compares (generally using RMSD calculations) the current pose to the one currently saved in the memory at each step. If they are too similar, the solution is rejected.

The Hybrid Docking Method¶
A hybrid strategy which combines different methods is often used in the hope of achieving improved solutions. They try to overcome the disadvantages of the individual methods and maintain their advantages. An example of Tabu search (TS) combined with genetic algorithms (GA) is illustrated below.

articles
Protein folding simulations of the hydrophobic-hydrophilic modelby combining tabu search with genetic algorithms Tianzi Jianga, Qinghua Cui, Guihua Shi, and Songde Ma J. Chem. Phys. 119 2003 10.1063/1.1592796
Methods for Incorporating Flexibility¶
Implementation of Flexibility into Docking Software¶
The rigid-body docking approaches presented in the previous section are often not sufficient to predict the structure of a protein complex from the separate unbound structures. Strategies incorporating molecular flexibility in the docking algorithm emerged in the early 1990s and are presented in the following pages.

Degrees of Freedom in Flexible Docking¶
The incorporation of molecular flexibility into docking algorithms poses huge challenges because of the need to add conformational multiplicity within an already difficult optimization problem of translational, and rotational degrees of freedom. Approximation algorithms need to be introduced to reduce the dimensionality of the problem and produce acceptable results within a reasonable computing time.
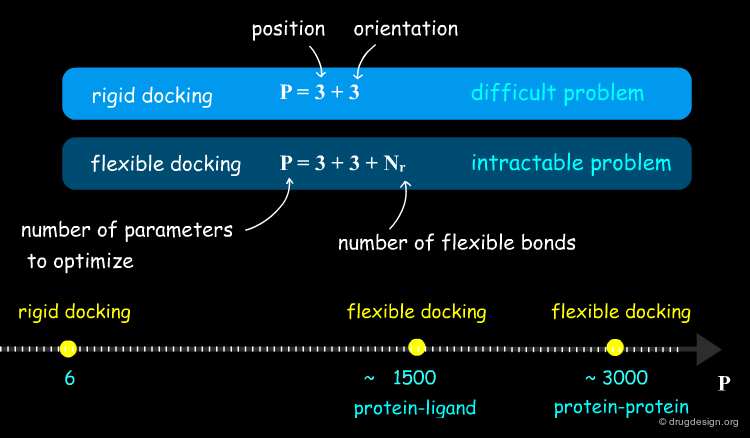

Possible Classification of Methods for Flexibility¶
There are four main ways of classifying docking methods by flexibility: (1) explicitly or implicitly: is flexibility treated explicitly by exploring the space of all possible conformations or implicitly by allowing some interpenetration between the molecular atoms? (2) flexibility type: side-chain movements, hinge-bending domain movements, loop movements, small molecule flexibility and others. (3) flexible molecules: protein, ligand, or both? (4) the stage of the method that accounts for flexibility: search, scoring or a separate refinement procedure.

Classification of Methods¶
In the following pages we first introduce the methods dealing with ligand flexibility; then we present how flexibility is introduced in proteins. Historically, ligand flexibility was first addressed in the 1990s by cheminformatic experts involved in drug discovery, whereas protein flexibility was recently addressed by university groups involved in structural biology and structural bioinformatics.

Incorporating Small Molecule Flexibility¶
The first attempt to accommodate some flexibility in biomolecules was to treat small molecules as flexible entities with torsional freedom and conformational multiplicity. This approach started in the mid nineties and was an important step towards the incorporation of protein flexibility.

Modeling Small Molecules as Flexible Entities¶
Handling small molecule conformational and torsion angle variability can reveal particular orientations and conformations that optimally bind to the protein. Favorable interactions (such as hydrogen bonds and hydrophobic interactions) make the difference between poorly active and potent compounds.
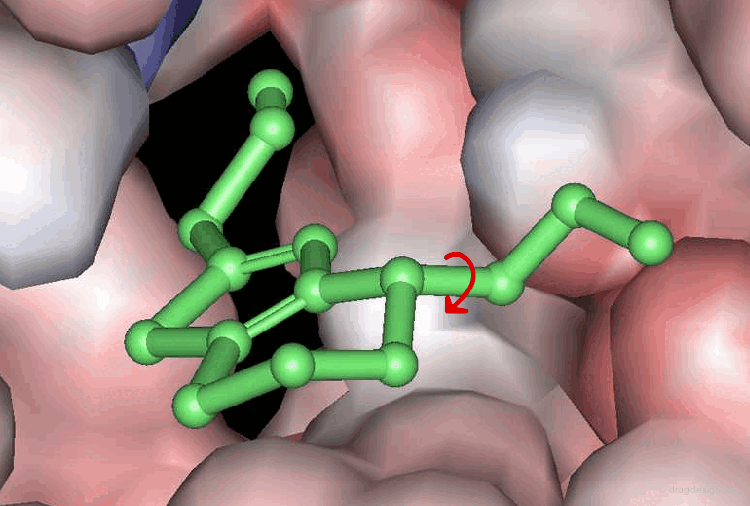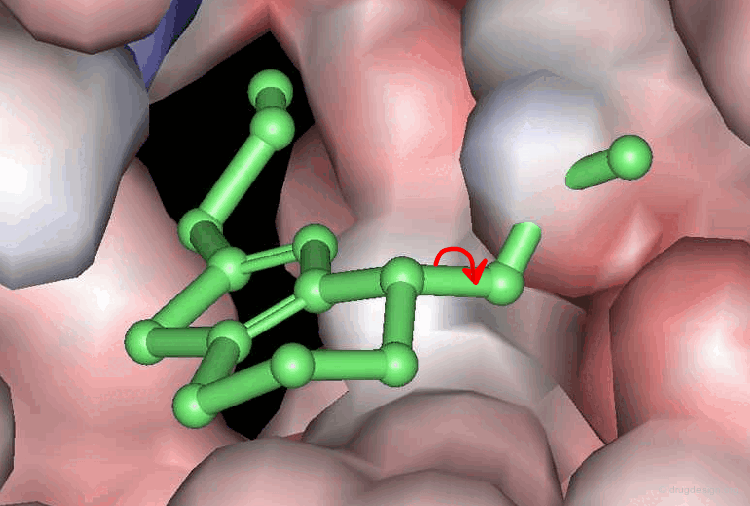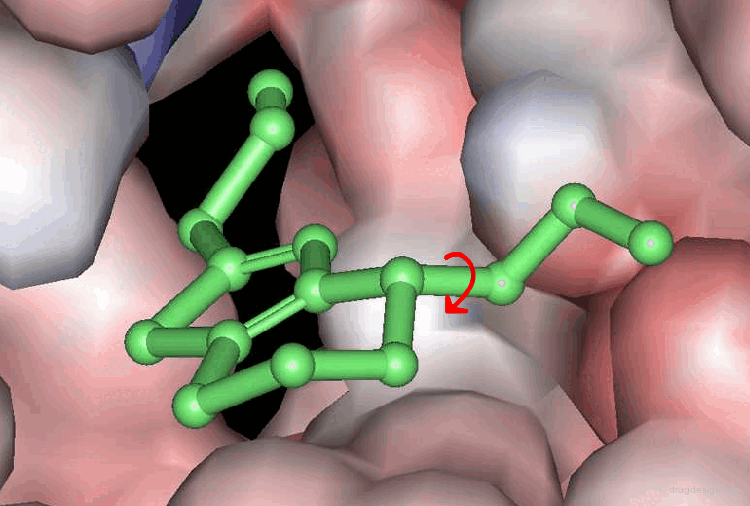
Small Molecule Flexibility¶
The treatment of small molecules capable of adopting multiple conformations is not a new idea; it emerged in the early 1980s with the first conformational analysis softwares incorporating force-field calculations. These aimed at studying the conformational potential surface of small molecules and acquiring insights into properties associated with their 3D features. This effort paved the way for tackling flexible docking of small molecules.
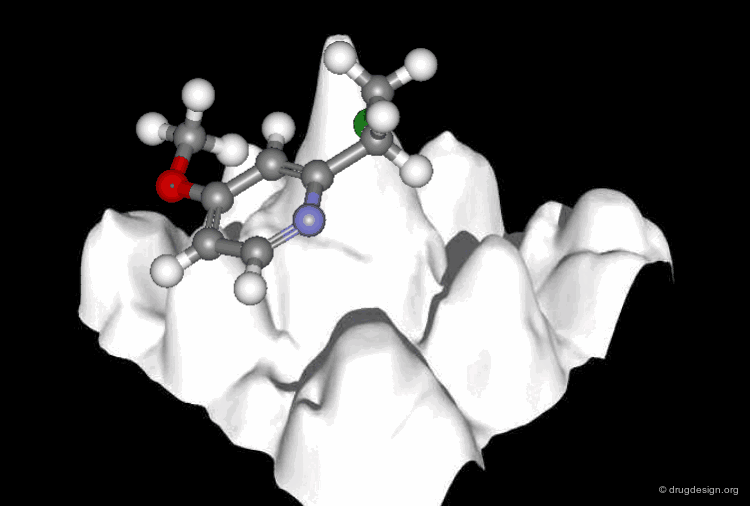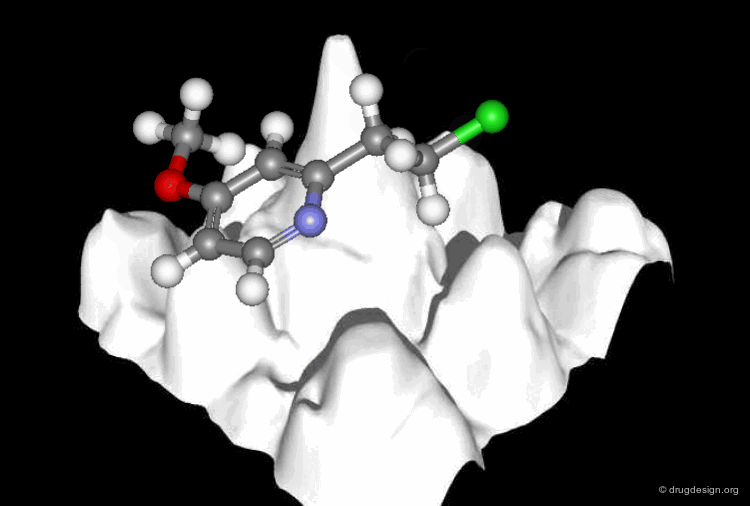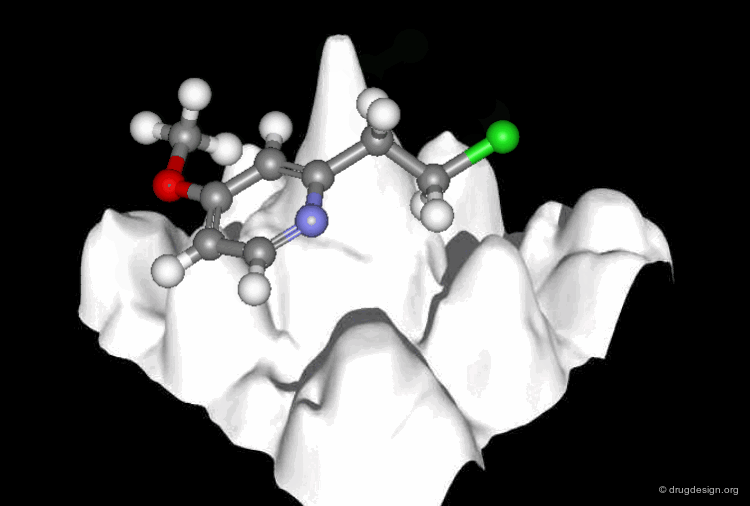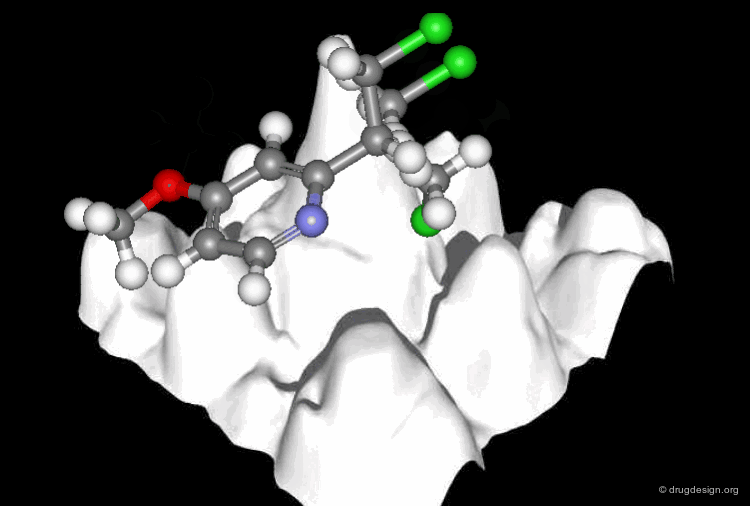
articles
Flexible ligand docking using conformational ensembles D. M. Lorber and B. K. Shoichet Protein Science 7 1998
Integration of Ligand Flexibility and Protein Structure¶
In the 1990s, the explosion of structure determination by X-ray crystallography prompted the integration of the conformational knowledge acquired about small molecules with the experimental data generated on proteins. This led to the development of the first flexible docking software and was the basis for a whole new discipline known as structure-based design.

Methods for Handling Ligand Flexibility Explicitly¶
Many methods have been developed for incorporating flexible small molecules into docking software; they include: (1) the ensemble docking method; (2) the fragmentation method and (3) the stochastic conformational search method. All three are presented in the following pages.

The Ensemble Docking Method¶
The simplest method to account for small molecule flexibility is to consider it as an "ensemble" of rigid and independent conformations. In the first step low energy conformers are generated. In the second step, rigid docking is applied for each conformer independently in order to find the most favorable small molecule-protein complex.

articles
A Geometric Approach to Macromolecule-Ligand Interactions Kuntz I. D., Blaney J. M., Oatley S. J., and Langridge R. L. J. Mol. Biol. 161 1982
Advantage of the Ensemble Docking Method¶
The advantage of the conformation ensemble approach is its ease of implementation since the rigid docking software does not need to be modified. Rigid docking programs (such as DOCK) were used for more than a decade (1982-1993) to dock sets of conformers manually. In the first step relevant conformations of the flexible small molecule are generated by conformational analysis programs and, in the second step these conformations are manually fed into a rigid docking program.

articles
A Geometric Approach to Macromolecule-Ligand Interactions Kuntz I. D., Blaney J. M., Oatley S. J., and Langridge R. L. J. Mol. Biol. 161 1982
The FLOG Software¶
The FLOG (Flexible Ligands On a Grid) software was the first docking program specially designed to assist the computational chemist in the repetitive task of docking. In phase one, FLOG generates low energy conformers of ligands stored in a database (using a distance geometry approach) then, each conformer is automatically assessed by a docking module. The authors demonstrated the feasibility of their method by docking methotrexate to dihydrofolate reductase.
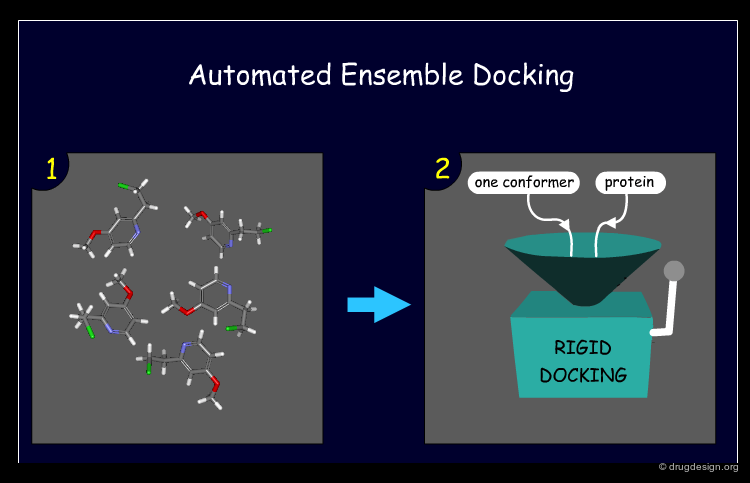
articles
FLOG: A System to Select 'Quasi-Flexible' Ligands Complementary to a Receptor of Known Three-Dimensional Structure Miller M. D. et al. J. Comput. Aided Mol. Des. 8 1994 10.1007/BF00119865
Flexibases: A Way to Enhance the Use of Molecular Docking Methods. Kearsley S. K. et al. J Comput-Aided Mol. Des. 8 1994
Problem of the Ensemble Docking Approach¶
The main problem with the ensemble docking method is that the computing time increases linearly with the number of conformers at hand. In order to limit computing to a reasonable time, the FLOG software generates up to 25 conformers per molecule that are selected using RMSD clustering.
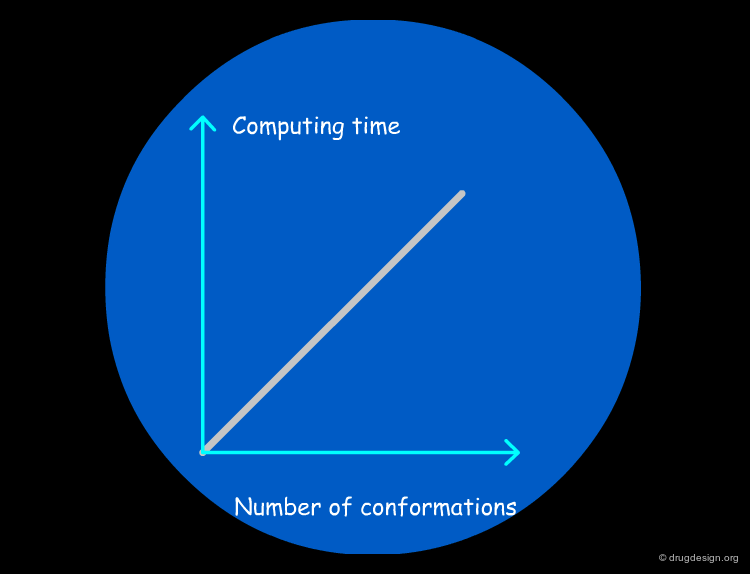
wikipedia
The Improved Ensemble Docking Method¶
In the ensemble docking method, many conformations are submitted to independent treatments; however, there are portions of the molecules that are identically oriented and docked repeatedly, a redundancy that could be avoided. In 1998 Lorber and Shoichet introduced an improved ensemble approach which removes the redundancy of docking multiple conformations, and dramatically lowers computational costs.

articles
Flexible ligand docking using conformational ensembles D. M. Lorber and B. K. Shoichet Protein Science 7 1998
Hierarchical Docking of Databases of Multiple Ligand Conformations David M. Lorbera and Brian K. Shoichet Current Topics in Medicinal Chemistry 5 2005
Flexible ligand docking using conformational ensembles LORBER D. M. and SHOICHET B. K. Protein Sci. 7 1998
Remove Redundancy in the Rigid Fragment¶
To reduce the redundancy in the docking alogrithm, Lorber and Shoichet broke down the small molecule into a rigid fragment (identical for all conformers, here in blue) and a flexible fragment. In order to remain in the same referential, all conformers are superimposed on the rigid moiety. The score of each conformer is equal to the sum of the score of the rigid part (calculated only once) and the score of the flexible part.
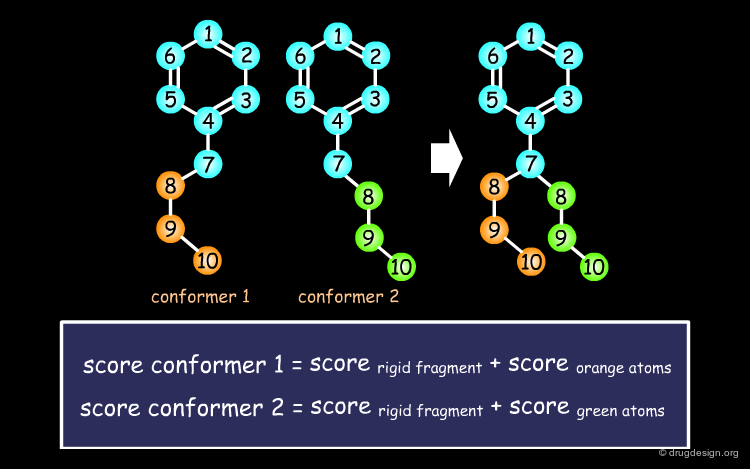
Remove Redundancy in the Flexible Fragment¶
Thanks to the efficient algorithm presented in the following pages, it is possible to remove redundancy in the flexible fragments as well. In the example below, only the atom colored in red corresponds to different conformations. Calculating for each conformer the interactions with the green atoms is a waste of time.
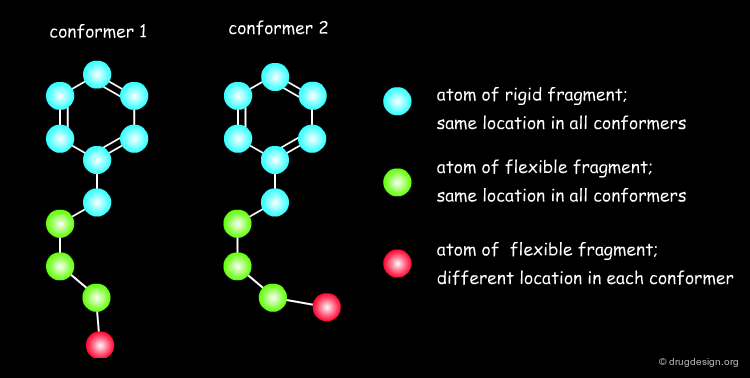
Score: Sum of Atom Interactions¶
Let us revisit the notion of scoring that constituted the foundation of the Lorber and Shoichet algorithm. The scoring of a small molecule ligand is the sum of the interactions of each of its atoms with those of the protein. In the figure below, if only the position of atom 3 (in yellow) is changed, only the interactions with this atom need to be recalculated, and not those with atoms whose location remain unchanged (in blue).

Step-1: Conformational Analysis¶
The Lorber and Shoichet algorithm consists of three consecutive steps. The first one is the generation of low energy conformers of the small molecule. This step is included in the FLOG program presented earlier, with the difference that it is not necessary to limit the total number of conformers to 25, because of the dramatic efficacy of the algorithm.
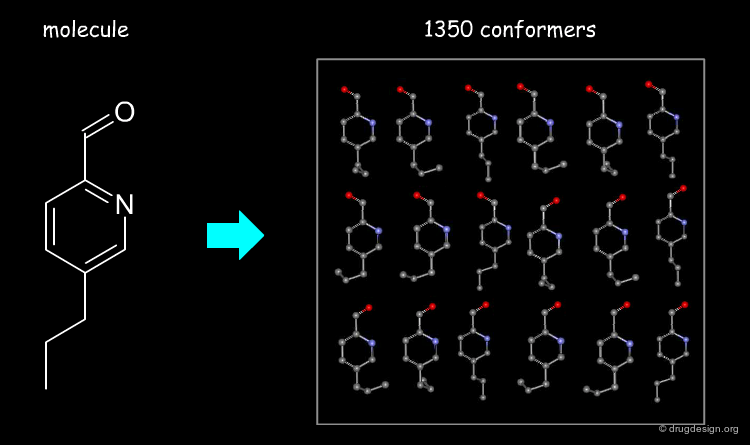
Step-2: Superimposition and Positioning¶
In the second step, all the conformers are superimposed onto the rigid fragment in order to place all atoms in the same reference frame. In the example below, A, B, C represent atoms of the flexible part; they can occupy 2, 3 or 9 different positions, respectively.


Step-3: Conformational Analysis¶
The key element of this new ensemble docking algorithm is the generation of a table which gives the atom positions for each conformer. For example conformer 1 is defined by atom A being in position 1; atom B being in position 1 and atom C being in position 1. For a given position of the rigid fragment, the score of the flexible part is assessed by calculating only for the atoms indicated in orange in the table. For example to score conformer 2, only the score for atom C needs to be computed since the scores for A1 and B1 were already calculated for conformer 1.
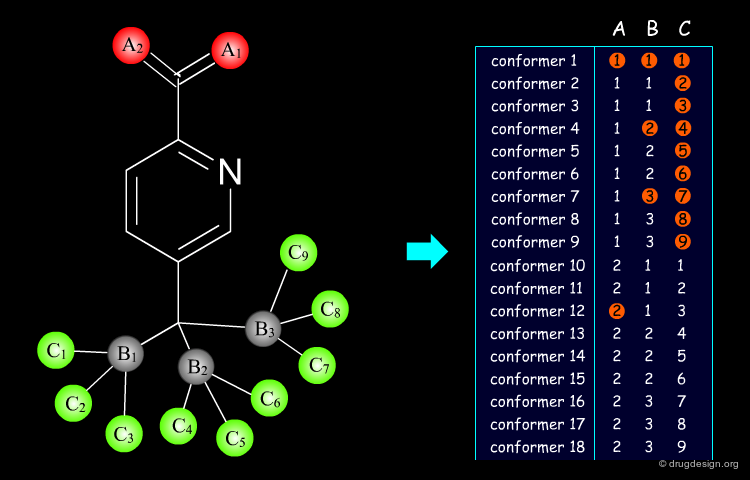
articles
Hierarchical Docking of Databases of Multiple Ligand Conformations David M. Lorber and Brian K. Shoichet Curr Top Med Chem 5(8) 2005
Dramatic Improvement in Computing Time¶
In the example on the previous page, instead of calculating 18x3=54 interactions for the flexible moiety, it is sufficient to calculate only 14 of them. As far as the rigid core is concerned, 18x8=144 interactions are calculated once and for all. The gain in computing time is appreciable, and this is extremely important because for each conformation we can have thousands of different poses and a library may contain millions of compounds.

articles
Hierarchical Docking of Databases of Multiple Ligand Conformations David M. Lorber and Brian K. Shoichet Curr Top Med Chem 5(8) 2005
Efficient Treatment of Clashes¶
In addition to the clever representation of atomic positions in the different conformers, the coordinates are also hierarchically organized in a tree to allow for rapid pruning of the branches when they clash with the protein. For example, for a given position of the rigid fragment, if atom B3 clashes with the receptor, atoms C7, C8 and C9 will not be assessed.

Validation of the Lorber-Shoichet Method¶
Lorber and Shoichet tested their method on two projects (dihydrofolate reductase and thymidylate synthase) using multiconformer databases of inhibitors. In both cases the binding modes of the molecules were calculated and proved to be close to those known experimentally. The method was 100-fold faster than docking the conformations independently. It could screen a database of 117,000 molecules represented by 34 million conformations in 1-4 CPU days on a workstation.

articles
Flexible ligand docking using conformational ensembles D. M. LORBER and B. K. SHOICHET Protein Science 7 1998
Extension to Analog Compounds¶
Recently Lorber and Shoichet extended their ensemble docking method to a library of molecules. The molecules are first grouped into families of related structures. Then all the members of every family are docked and scored using the same efficient strategy previously presented. This way proved to be of great practical utility since many molecules that are contained in databases (e.g. corporate databases) resemble one another. It turned the docking of large databases of flexible molecules common practice because it requires relatively modest computational resources.
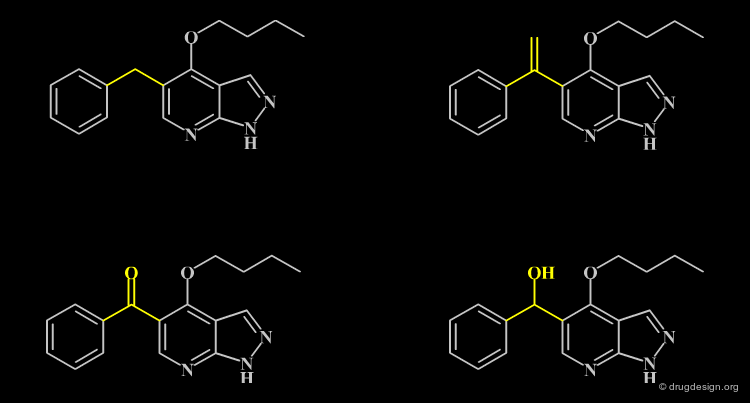
articles
Docking molecules by families to increase the diversity of hits in database screens: computational strategy and experimental evaluation Su A.I, Lorber D.M., Weston G.S., Baase W.A., Matthews B.W., Shoichet B.K. Proteins: Structure Function and Genetics 42(2) 2001 10.1002/1097-0134(20010201)42:2<279::AID-PROT150>3.0.CO;2-U
The Fragmentation Docking Method¶
The second class of docking methods handling small molecule flexibility is the fragmentation method. These methods break down the molecule into small rigid fragments. The fragments are then reassembled in the binding pocket. Two different approaches are used for reassembling the disconnected moieties: the place-and-join and the incremental approach. One of the advantages of fragmentation methods is that they enable efficient pruning of the search tree and early elimination of solutions that are not worth assessment.

articles
The computer program LUDI: A new method for the de novo design of enzyme inhibitors Boehm, H. J J. Computer-Aided Molecular Design 6 1992
Place-and-Join Algorithm¶
The place-and-join method splits the molecule into rigid subparts. Each subpart is docked independently. Assembly of the fragments is then done by looking at their relative location and assessing the possibility of re-connecting them with the connectivity of the initial molecule, in geometrically correct conformations.


articles
Docking Flexible Ligands to Macromolecular Receptors by Molecular Shape DesJarlais, R.L., Sheridan, R.P., Dixon, J.S., Kuntz, I.D., Venkataraghavan, R. J. Med. Chem. 29 1986
On the Use of Ludi to Search the Fine Chemicals Directory for Ligands of Proteins of Known Three-Dimensional Structure H.J. Bohm J. Comput. Aided Molec. Des. 8 1994
a program for rapidly producing pharmacophorically relevant molecular superpositions. M.D. Miller, R.P. Sheridan, S.K. Kearsley J. Med. Chem. 42 1999 10.1021/jm9806143
Principle of the Place-and-Join Method¶
The place-and-join method is based on the additivity hypothesis of atomic interactions between a protein and a small molecule, which allows local regions to be treated independently. A complicated problem (the docking of a big flexible molecule) is decomposed into a set of small problems (docking of small rigid fragments). The initial molecule is then regenerated by assembling the elementary docked fragments.
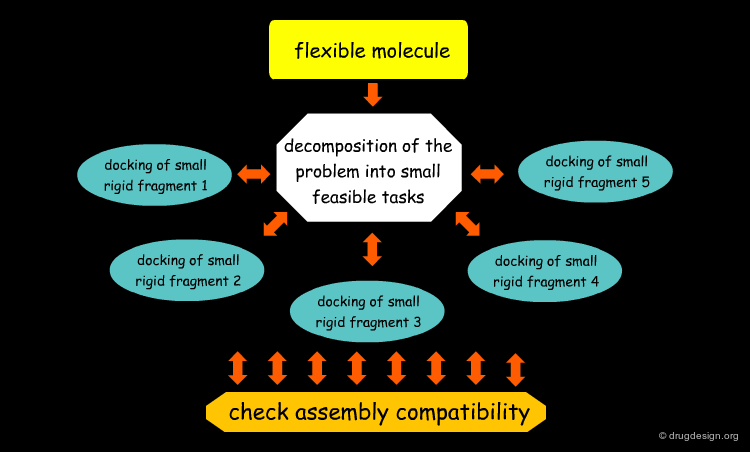
Difficulty of the Place and Join Method¶
The main difficulty of the place-and-join method is that many placements are generated for each individual fragment, so that going over all the possibilities in order to select best scoring feasible assemblies is computationally expensive; this requires an efficient management of the individual docked solutions.

Incremental-Based Methods¶
The incremental based approach is another alternative to the fragmentation method instead of docking all small molecule subparts independently in the binding site, it starts with an initial core docked in the active site, and new fragments are progressively added and minimized. The treatment is terminated when the entire molecule is formed.
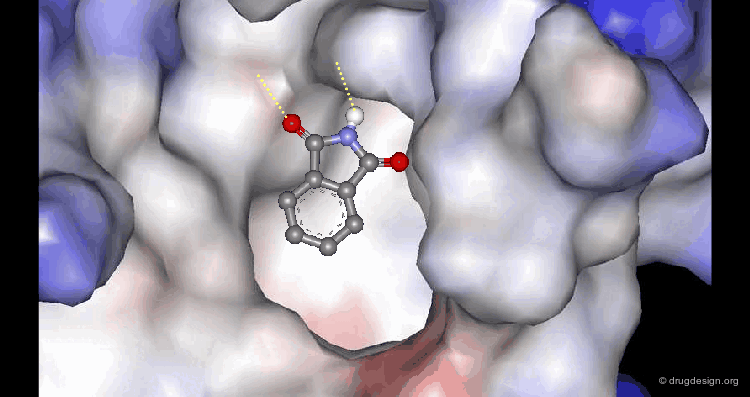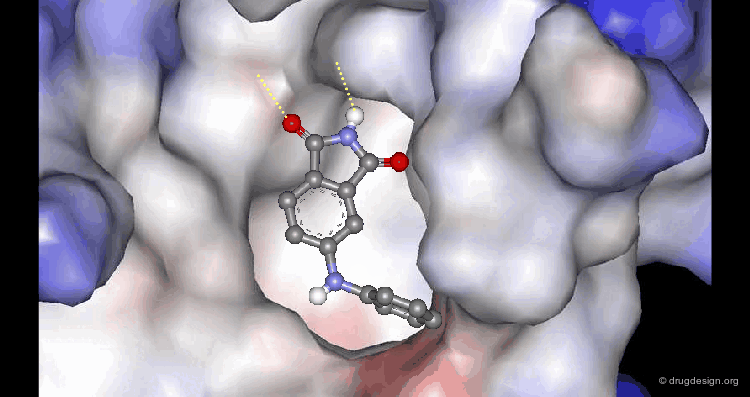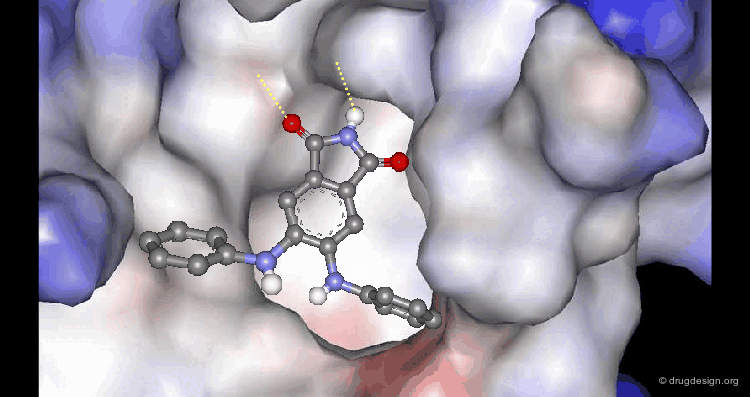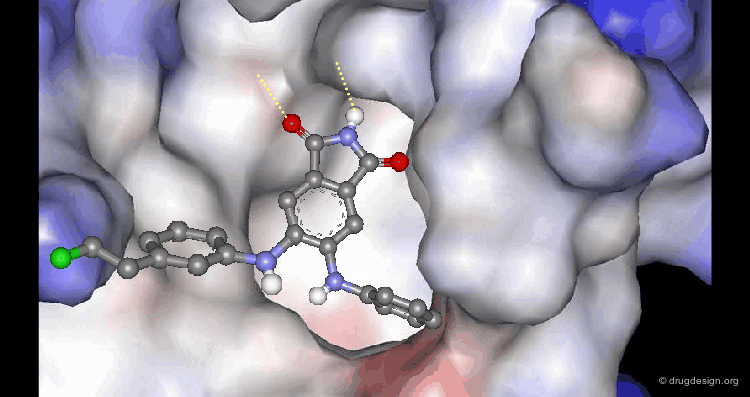
articles
A fast flexible docking method using an incremental construction algorithm Rarey, M.; Kramer, B.; Lengauer, T.and Klebe, G. J. Mol. Biol 261 1996
Incremental Algorithm¶
The main problem of incremental construction algorithms is their dependence on the choice of the first fragment to be docked, which requires an exploration of several possibilities. In the example below the initial rigid fragment has been docked three times (A, B, C). For each one, we have a set of potential solutions that can be generated. Each intermediate level is extended to the next one by adding a new next fragment in all possible configurations. Solutions with a bad score are eliminated.

Stochastic Search Methods¶
Stochastic search methods modify the conformation of the small molecule in the receptor site and assess it on the fly. These methods were already discussed in the rigid docking section. Flexibility introduces an additional degree of complexity. In the case of genetic algorithms, the torsion angles are added on the chromosomes; in Monte Carlo based methods they are set as parameters for optimization.

articles
Development and validation of a genetic algorithm for flexible docking Jones, G.; Willet, P.; Glen, R.; Leach J. Mol. Biol 267 1997
Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function Morris, G. M., Goodsell, D. S., Halliday, R.S., Huey, R., Hart, W. E., Belew, R. K. and Olson, A. J. Computational Chemistry 19 1998
ICM: a new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation Abagyan, R.; Totrov, M.; Kuznetsov, D. J. Comput. Chem. 15(5) 1994
Software package for protein modeling and docking Trosset, J.; Scheraga, H. J. Comput. Chem. 20 1999
GOLD Program¶
GOLD is an example of a program based on genetic algorithms. Each chromosome encodes information about the conformation of the ligand (torsion angles) and the hydrogen bond patterns between ligand and protein in order to maximize the number of inter-molecular hydrogen bonds for each GA move.
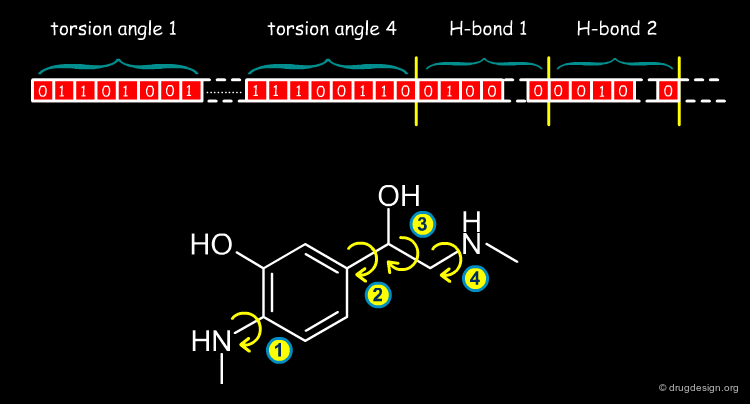
articles
Development and Validation of a Genetic Algorithm for Flexible Docking Jones, G.; Willet, P.; Glen, R.; Leach, A. J. Mol. Biol. 267 1997
Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation Jones, G.; Willet, P.; Glen, R. J. Mol. Biol. 245 1995
A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. Jones, G.; Willet, P.; Glen, R. J. Comput. Aid. Mol. Des. 9 1995
Incorporating Protein Flexibility¶
Incorporating protein flexibility into molecular docking software is a difficult optimization problem involving a huge number of degrees of freedom that represent the receptor flexibility, making this challenge not computationally feasible. In the last few years several practical approaches have been developed to tackle this problem, some of which will be presented in the following pages.



articles
The impact of protein flexibility on protein-protein docking Ehrlich, L.; Nilges, M.; Wade, R. Proteins: Structure Function and Genetics 58
book
M. Teodoro and G. Phillips and L. Kavraki IEEE International Conference on Robotics and Automation IEEE Press 2001
Importance of Modeling Protein Flexibility¶
In 1999 Murray et al. demonstrated the shortcomings of the protein rigid docking approach by carrying out rigid "cross-docking" experiments for three enzymes: thrombin, thermolysin and the influenza virus neuraminidase, based on all complexes available for each enzyme, where each ligand was docked into all the proteins. They found that in 51% of the cases the program failed to dock the small molecule correctly. This study highlights the importance of modeling protein flexibility in computational docking.

articles
The sensitivity of the results of molecular docking to induced fit effects: Application to thrombin, thermolysin and neuraminidase. Murray, C. W.; Baxter, C. A.; Frenkel, A. D. J. Comput.-Aided Mol. Des. 13 1999 10.1023/A:1008015827877
Historical Note¶
Computational approaches addressing protein mobility for docking purposes emerged in the early 1990s. The first approaches started with the implicit soft docking method, followed by the use of discrete side chain rotamer libraries. Backbone and domain motions have recently started to be tackled in docking simulations. These methods are presented in the following pages.

Flexibility Through Soft Scoring Functions¶
The first approach to tackle the protein flexibility problem is the 'soft docking' method introduced by Jiang et al. in 1991; it allows for slight penetrations between the receptor and the ligand molecules. This is a mathematical trick where the receptor and the ligand are held rigid and a 'soft' scoring function is used, allowing some overlap between them.
articles
Soft Docking: Matching of Molecular Surface Cubes. Jiang, F.; Kim, S. J. Mol. Biol. 219 1991
BiGGER: a new (soft) docking algorithm for predicting protein interactions Palma, P.N., Krippahl, L., Wampler, J.E., and Moura, J.J.G. Proteins: Structure Function and Genetics 39 2000 10.1002/(SICI)1097-0134(20000601)39:4<372::AID-PROT100>3.0.CO;2-Q
Reduce the Importance of Steric Clashes¶
In the example below, the fit between the substrate and the protein is acceptable, except however for a bump with the phenyl group. The idea behind a soft scoring function is that instead of resolving bumps by conformational changes, it is possible to reduce their importance by using softer interaction energy functions. A soft scoring function increases the chances of not overlooking good solutions.
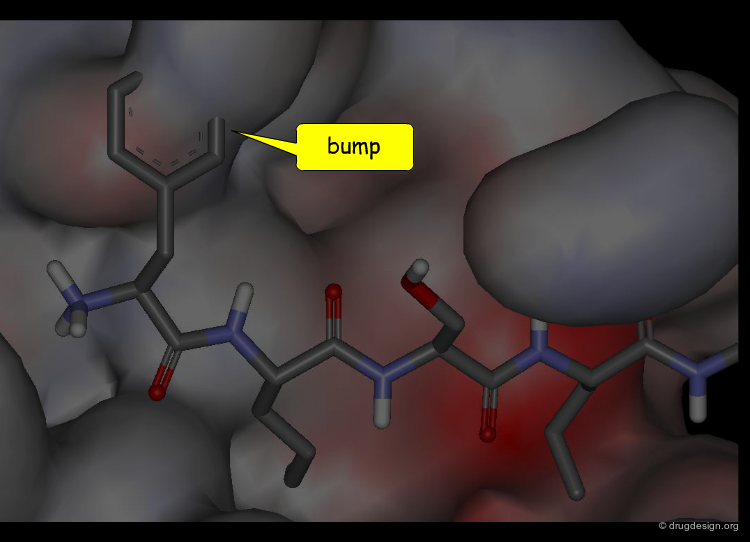
Soft Van der Waals Repulsion Functions¶
When calculated with force fields from molecular mechanics, steric clashes correspond to high energies. Modifying the normal Van der Waals repulsion function (in blue) into a softer curve (such as the yellow one) enables them to be less dominant and to simulate the plasticity of the receptor without changing its geometry.

Decreasing Van der Waals Radii¶
Another way to reduce the importance of steric clashes is to use reduced radii for the atoms. In the example below, bumps and their associated energy penalties are considerably reduced by altering the van der Waals radii of the ligand.
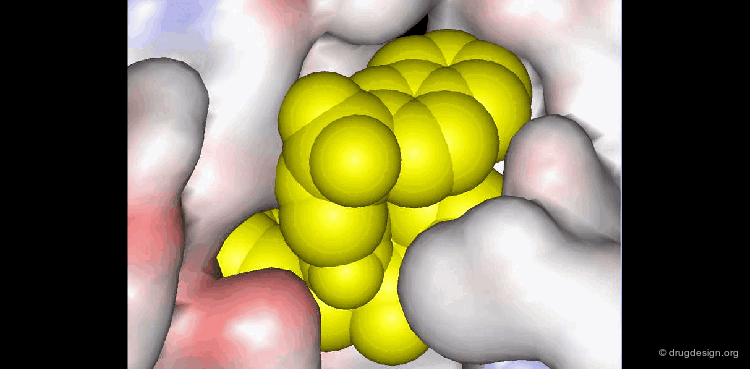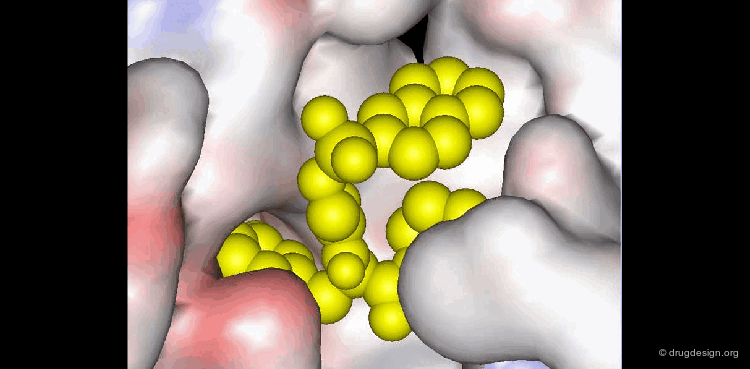

wikipedia
Soft Electrostatic Repulsion Potentials¶
The same principles apply for repulsive electrostatic interactions where the theoretical energy function (in blue) can be softened by a less drastic curve used at short interaction distances (in yellow).

Soft Scoring Functions in Protein-Protein Docking¶
Protein-protein docking is well suited for using the soft scoring method, where complementary surfaces between both proteins are generally huge thus increasing the chance to miss a docking configuration due to clashing atoms.

wikipedia
Implicit Flexibility in Protein-Protein Docking¶
In protein-protein docking where shape complementarity is computed as the number of overlapping surface grid voxels, "soft scoring" is done by allowing some contacts with interior voxels as well. The contacts with interior voxels are penalized, but the solution is not disqualified.

articles
Anchor residues in protein-protein interactions Rajamani, D.; Thiel, S.; Vajda, S.; Camacho, C Proc. Natl. Acad. Sci. USA 101 2004 None
wikipedia
Problems with Soft Scoring¶
The major disadvantage of the soft scoring approach is the large number of false-positive hits it produces which therefore makes it harder to discriminate the near native solution from the other candidates.

wikipedia
Soft Scoring as a First Filtering Method¶
It has been suggested that the soft scoring function approach should be used as a first filtering algorithm to be complemented by more refined quantitative methods.
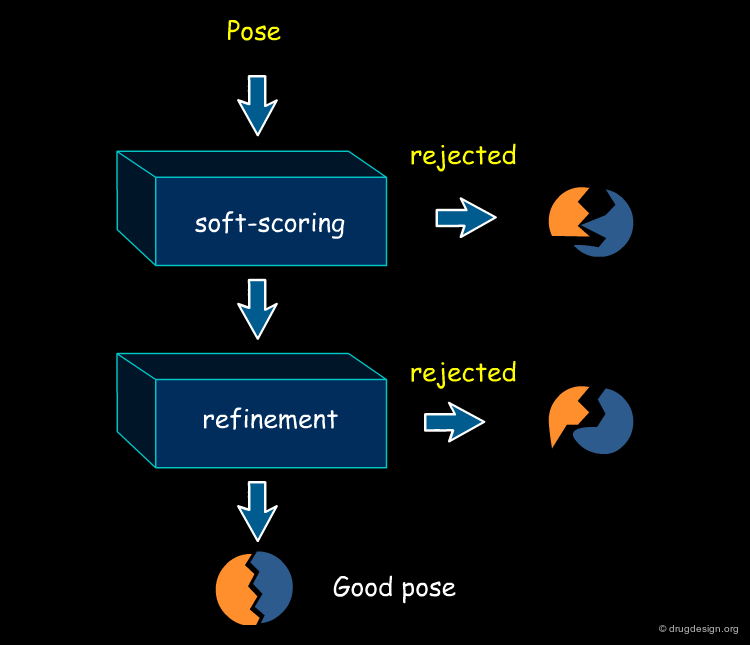
Protein Side-Chains Flexibility¶
Side chains are critical for the binding of the ligand to proteins. In the past the incorporation of side-chain flexibility in docking algorithms was hampered because of the great number of possible conformations. New methods were recently introduced allowing conformational changes of side chain residues while keeping the protein backbone rigid, and this is one step towards the simulation of the full flexibility of the protein.
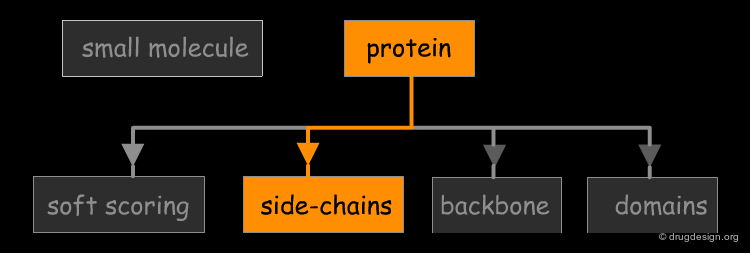

articles
Accommodating protein flexibility in computational drug design Carlson HA, McCammon JA. Mol Pharmacol. 57(2) 2000
book
Importance of Modeling Side-Chain Mobility¶
Altering the side chain conformations in docking calculations enables the maximization of favorable interactions with the protein. Studies comparing the X-ray structures of complexes with that of the corresponding unbound proteins (in protein-protein and protein-ligand associations) reveal that about 60% of side chains modify their conformations upon binding. They demonstrate the importance of modeling side-chain flexibility in computational docking.
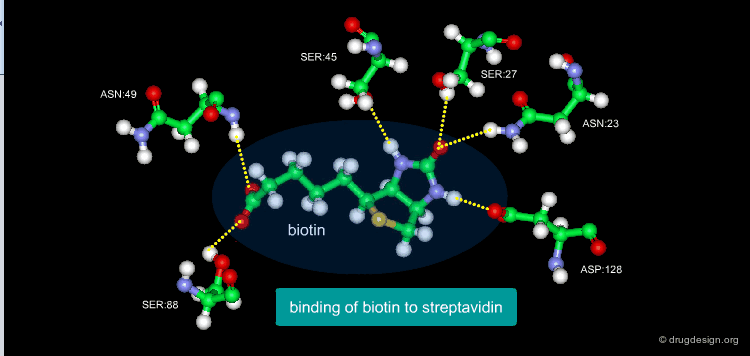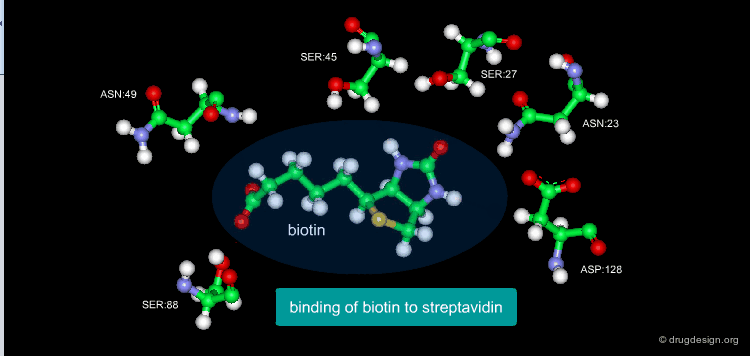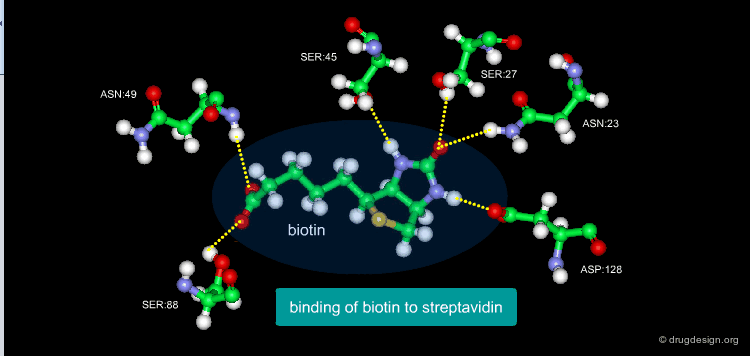
articles
An analysis of conformational changes on protein-protein association: Implications for predictive docking Betts, M.J. and Sternberg, M.J. Protein Eng. 12 1999
Side-chain flexibility in proteins upon ligand binding Najmanovich, R., Kuttner, J., Sobolev, V., and Edelman, M. Proteins 39 2000
Side-chain flexibility in protein-ligand binding: The minimal rotation hypothesis Zavodszky M.I. Protein Science 14 2005 10.1110/ps.041153605
A combinatorial approach to protein docking with flexible side chains Althaus, E.; Kohlbacher, O.; Lenhof, H.; Mueller, P J. Comp. Biol 9 2002
Determine the Optimum Combination of Side-Chains¶
The goal of flexible side-chain algorithm is to determine an optimum combination of side-chain conformations of residues interacting with the ligand (in yellow in the view) for a given backbone geometry and a given orientation of the ligand.

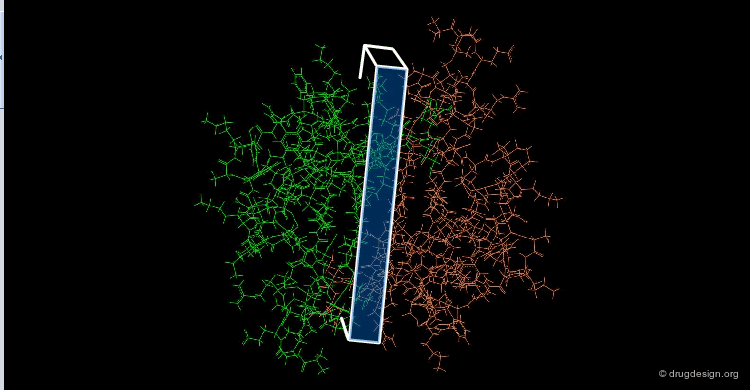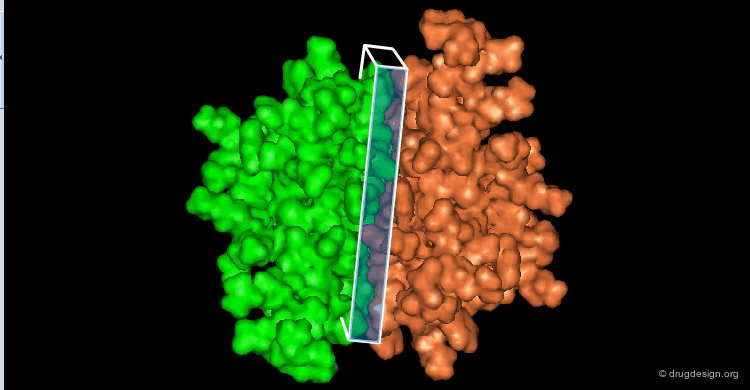
Combinatorial Explosion¶
Exploring the conformational space of protein side chains is a complex optimization problem that leads to a combinatorial explosion of conformers. For example, if we consider a typical binding site with 20 side-chains, the total number of possible rotamer combinations can become as high as 1020. Assessing side-chain conformations of a protein is unfeasible and impractical.
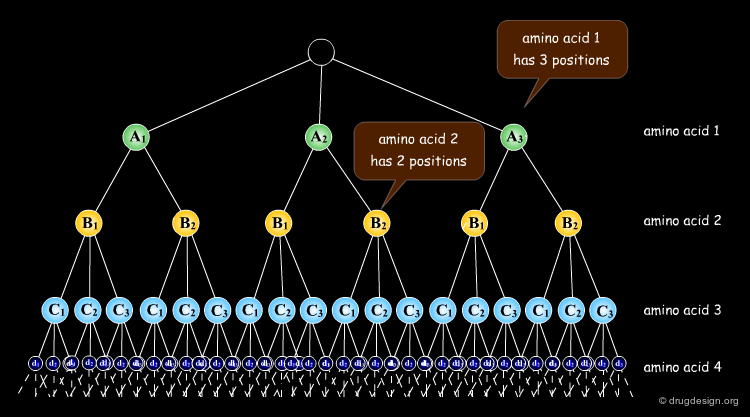
Side Chain Rotamer Libraries¶
In 1987 Ponder and Richards show that protein side chains can be represented adequately by a small set of discrete rotamers. In their studies they analyse side-chain Ramachadran plots of pdb structures. They show that 17 of the 20 amino acids can be represented adequately by 67 side-chain rotamers. Additionally, backbone-dependent rotamer libraries became available: the discretization of the conformational space greatly simplified the problem and enabled side-chain flexiblity to be tackled.


articles
Rotamer libraries in the 21st century Dunbrack RL Jr. Curr Opin Struct Biol. 12(4) 2002
Tertiary templates for proteins - use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Ponder and F. Richards. J. Mol. Biol. 193 1987
Ligand docking to proteins with discrete side-chain flexibility Leach A.R. J. Mol. Biol. 235 1994 10.1016/S0022-2836(05)80038-5
Media
The rotamers or GLU PHE and ARG are from the Rotamer kinemage library Rotamer Kinemages Library
From Folding to Docking¶
The side-chain combination problem has been first addressed in protein folding to assign optimal side chain orientations for a protein constructed by homology modeling. In 1994 Leach introduced the side chain flexibility idea into docking algorithms.
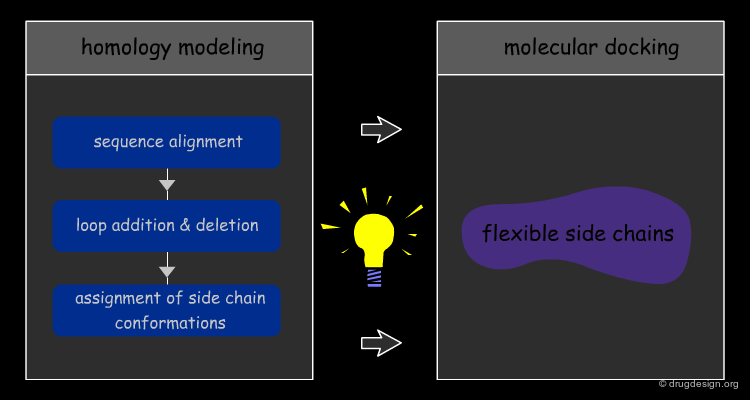
articles
Ligand docking to proteins with discrete side-chain flexibility Leach A.R. J. Mol. Biol. 235 1994 10.1016/S0022-2836(05)80038-5
Exploring the Conformational Space of Protein Side Chains Using Dead-End Elimination and the A* Algorithm Leach A.R. et al. Protein 33 1998 10.1002/(SICI)1097-0134(19981101)33:2<227::AID-PROT7>3.0.CO;2-F
wikipedia
The Leach Algorithm¶
Leach explored the side chain conformation space using dead-end elimination (DEE) algorithms and successfully avoided the problem resulting from the combinatorial explosion of the side chain rotamers. The basic idea of dead-end elimination algorithms is to prune rotamers that cannot be members of the global minimum energy conformation.

articles
Ligand docking to proteins with discrete side-chain flexibility Leach A.R. J. Mol. Biol. 235 1994 10.1016/S0022-2836(05)80038-5
The dead-end elimination theorem and its use in protein side-chain positioning Johan Desmet et al. Nature 356 10.1038/356539a0
wikipedia
Generation and Minimization of Complexes¶
In a study designed to predict the structural effects on HIV-1 protease mutant complexes, Schaffer et al. enhanced the previous docking method by combining the dead-end elimination algorithm with minimizations. In the first step, models of the protein with all the required conformations are generated. In step two, energy minimizations are made on a reduced set of atoms to find the most favorable ligand-protein complex.
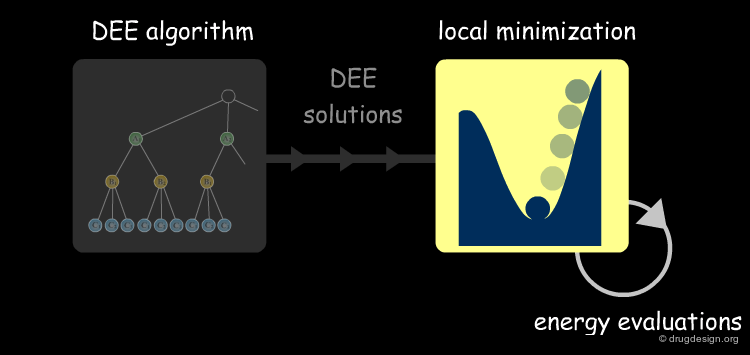
articles
Predicting structural effects in HIV-1 protease mutant complexes with flexible ligand docking and protein side-chain optimization Schaffer L and Verkhivker GM Proteins: Structure Function and Genetics 33(2) 1998 10.1002/(SICI)1097-0134(19981101)33:2<295::AID-PROT12>3.0.CO;2-F
Other Optimization Methods¶
The best side-chain combination problem has been approached by many different methods. Ehrlich combines rigid body and torsion angle dynamics. Zacharias uses a reduced protein representation to account for the side-chain flexibility. RosettaDock places side-chains using a simulated annealing algorithm; other methods add rotamer selection to the genetic algorithm or Monte Carlo optimization. More recently Althaus et al. introduced the multi-greedy and the branch-and-cut methods.

articles
Protein-protein docking with a reduced protein model accounting for side-chain flexibility MARTIN ZACHARIAS Protein Science 12 2003 10.1110/ps.0239303
The Impact of Protein Flexibility on Protein-protein Docking Ehrlich et al. Proteins 58 2005 10.1002/prot.20272
Rapid refinement of protein interfaces incorporating solvation: Application to the docking problem Jackson, R.; Gabb, H.; Sternberg, M J. Mol. Biol 276 1998
Protein-Protein Docking with Simultaneous Optimization of Rigid-body Displacement and Side-chain Conformations
JMB 331 2003 10.1016/S0022-2836(03)00670-3
wikipedia
Restricting Searches and Minimizations¶
Incorporation of side-chain flexibility in docking algorithms is computationally very expensive. Strategies used to reduce the molecular search space are presented in the following pages.

Identify Key Residues for the Interaction¶
In many protein-protein interactions the concavity of one protein enters the cavity of the other. The concave region may include a small number of highly exposed long and very flexible side chains like lysine or arginine. In this case the flexibility in the cavity is very limited. These side chains are the "key" recognition residues that fit into the cavity of the receptor in very specific conformations and can serve to restrict the conformational search.

Restrict the Search to Exposed Side Chains¶
Another strategy for reducing the conformational complexity introduced by the side-chains consists of restricting the search to highly exposed side-chains of the protein, because buried and partially solvent exposed residues do not contribute to the binding. It has been estimated that about 60% of the surface residues are highly exposed.

Backbone and Side Chain Flexibility¶
One of the greatest challenges in molecular docking is the incorporation of backbone and side chain flexibility in docking algorithms. Several studies suggest that local backbone movements affect the dynamics of complex formation, hence including this type of flexibility is important for successful docking prediction. Recently, there have been several attempts to include these movements in docking.
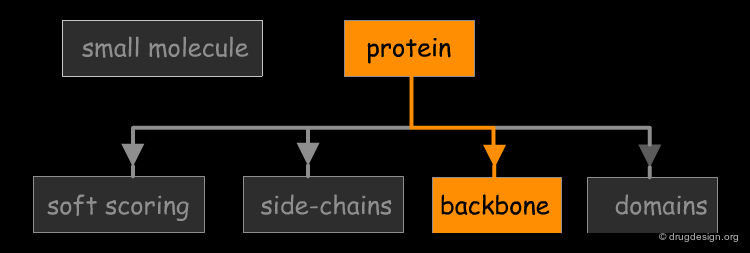

articles
Accommodating protein flexibility in computational drug design Carlson HA, McCammon JA. Mol Pharmacol. 57(2) 2000
Conventional Methods not Adapted¶
Due to the complexity level introduced by the huge number degrees of freedom, traditional methods such as the systematic approach which aims at checking all possible combinations or stochastic algorithms cannot be used as a general method.
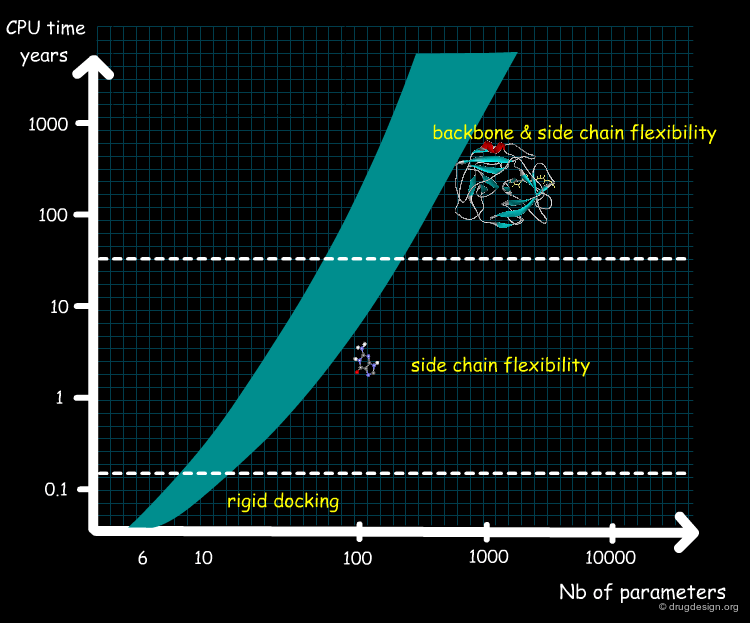
The Multiple Protein Structure (MPS) Approach¶
The most efficient method for considering the full flexibility of the protein that can be applicable to the screening of large libraries of ligands is the "Multiple Protein Structure" (MPS) approach. Other names are sometimes given to the approach and include the following: "multiple receptor structure"; "ensemble approach" and "multiple copy approach". This method is presented in the following pages.

articles
Representing receptor flexibility in ligand docking through relevant normal modes Cavasotto, C.; Kovacs, J.; Abagyan, R. J. Amer. Chem. Soc 127 2005
Docking macromolecules with flexible segments Bastard, K.; Thureau, A.; Lavery, R.; Prevost, C J. Comput. Chem 24 2003
A hybrid method of molecular dynamics and harmonic dynamics for docking of flexible ligand to flexible receptor Tatsumi, R.; Fukunishi, Y.; Nakamura, H J. Comput. Chem 25 2004
Principle of the MPS Approach¶
The Multiple Protein Structure approach is based on the use of multiple structures of the target protein as obtained either from experimental determinations or generated by theoretical simulations. The example below illustrates the CDK2 protein as determined by seven independent X-ray studies.

Media
This picture was done using MATRAS Multiple 3D Alignment tool MATRAS (Protein 3D Structure Comparison)
Sources of Multiple Protein Structures¶
The simplest way to obtain a multiple protein structure (MPS) is to use different crystallographic or NMR structures available for a protein. Alternatively, MPS can be generated using computational methods such as molecular dynamics simulations, Monte Carlo simulations or normal mode calculations. Predictive methods generally start from a single structure determined experimentally.

articles
Analysis of domain motions by approximate normal mode calculations Hinsen, K. Proteins: Structure Function and Genetics 33 1998
Predictions of Protein Flexibility: First Order Measures Kovacs, J.; Chacon, P.; Abagyan, R. Proteins: Structure Function and Genetics 56 2004
Protein Flexibility Predictions Using Graph Theory Jacobs, D.; Rader, A.; Kuhn, L.; Thorpe, M Proteins: Structure Function and Genetics 44 2001
Computational Drug Design Accommodating Receptor Flexibility: The Relaxed Complex Scheme Lin, J.; Perryman, A.; Schames, J.; McCammon, J J. Amer. Chem. Soc 124 2002
MPS: a Good Model for the Recognition Process¶
A protein exists as a number of conformational substates that constantly jump from one substate to the other; thus the MPS is a good representation of its conformational multiplicity. Ideally, each protein of the MPS should represent a funnel in the energy landscape; it is therefore important to have a good sampling in the MPS. It has been shown that the MPS obtained experimentally generally produces a better sampling of the protein energy profile than that obtained by predictive methods.

How the MPS are Exploited?¶
After having obtained a multiple protein structure (MPS), we now need to see how the MPS can be exploited for practical docking applications. Diverse approaches have been developed in recent years and in the following pages we present some of them.
articles
Protein Flexibility is an Important Component of Structure-Based Drug Discovery Carlson H. A. Current Pharmaceutical Design 8 2002 10.2174/1381612023394232
Successive and Independent Docking Treatments¶
The most trivial method of exploiting the MPS is to treat each member of the MPS independently, by applying a rigid docking for each protein of the ensemble. Ligands that bind consistently well to many proteins of the MPS correspond to good solutions. The advantage of this approach is its ease of implementation since the rigid docking software does not need to be modified.

# Acetylcholinesterase Example¶
Pang and Kozikowski used this simple method for the first time to study the binding of Huperzine-A to Acetylcholinesterase (AChE). A short molecular dynamic simulation of AChE was used to sample 69 conformations of the protein. Huperzine-A was then rigidly docked on each of these structures. The study correctly predicted where and how Huperzine-A binds to AChE.

articles
Prediction of the binding sites of huperzine A in acetylcholinesterase by docking studies Yuan-Ping Pang and Alan P. Kozikowski Journal of Computer-Aided Molecular Design 8 1994 10.1007/BF00124014
Media
This picture was made using the RCSB PDB Ligand Explorer 2.0 (Powered by the MBT) Launch PDB Ligand Explorer
The United Protein Approach¶
A more efficient method was developed by Claussen et al. (program FlexE) and consists of combining the multiple receptor conformations into a united protein description obtained from the superimposition of all the structures of the ensemble. The "united protein description" defines any virtual protein conformation formed by the combinatorial arrangement of any part of the original ensemble.

articles
FlexE: efficient molecular docking considering protein structure variations. Claussen H. et al. J Mol Biol 308(2) 2001 doi:10.1006/jmbi.2001.4551
# Key Concept of FlexE¶
FlexE considers the induced-fit locally, each residue (backbone included) in the united protein structure is independent of the others. When a new candidate molecule is docked, the best scoring combination for each residue is selected. In this way new protein conformations can be generated.

# Remove Redundant Information¶
In the first stage of the method the multiple structures are superimposed, similar rotamers are merged, dissimilar ones are kept as alternatives, they are called "instances". A clustering algorithm is applied to remove multiple instances of the same rotamer. This step of removing redundant information is by itself a massive improvement as compared to the initial method presented earlier.

# FlexE: Incompatibility Graph¶
A key element of the FlexE algorithm is the construction of an incompatibility graph which describes instances that should not be combined together in a same protein. Two instances are considered incompatible if: (1) they belong to the same side-chain; (2) they overlap with each other or (3) the backbone connectivity is not preserved. In the figure below each node represents one instance; nodes connected together are incompatible. A valid protein structure corresponds to an independent set (disconnected sub-graph) in the incompatibility graph.

# FlexE: Search & Scoring¶
In the docking stage we are looking for a valid protein that best interacts with the ligand (highest score). The problem of finding independent sets in the graph is similar to clique detection.
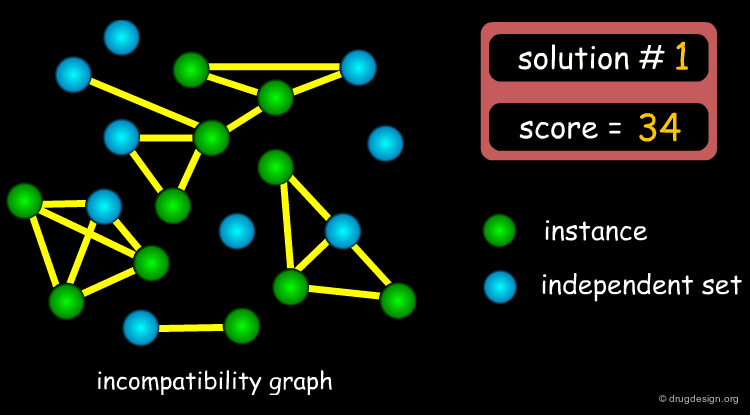
The Average Grid Approach¶
The average grid approach is another method that exploits MPS. A grid is used to characterize the shape and 3D specificity of each protein of the ensemble. The space is systematically explored by calculating the interaction energy (generally Van der Waals and electrostatic energies) between a chemical probe and each protein structure, at each grid point.

articles
Molecular docking to ensembles of protein structures Molecular docking to ensembles of protein structures Knegtel, R.; Kuntz, I.; Oshiro, C. J. Mol. Biol 266 1997
Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in Auto-Dock. Osterberg, F.; Morris, G.; Sanner, M.; Olson, A.; Goodsell Proteins: Structure Function and Genetics 46 2002
# Single Grid Combining MPS Information¶
The multiple structures are aligned in 3D; the main idea is to combine information from multiple structures in one single grid and to use it to guide the docking of a ligand. In this complementary model of the receptor, the information from aligned multiple protein structures is combined into "energy-weighted" averages of the interaction energies and "geometry-weighted" averages.

# Scoring Tolerance with MPS-based Grids¶
The grids based on multiple protein structures provide a way for more tolerant scoring of docking candidate transformations. Information from different sources of protein conformational flexibility can be combined.

# Average Grid Approach vs. Soft Scoring¶
The average grid approach can be considered as an improvement on soft-scoring function calculations. Both methods alter the scoring function to allow for slight penetrations between the interacting molecules. In the average grid approach, information from the MPS results from many local scoring functions, depending on the flexibility regions of the protein; whereas in the soft-docking approach a mathematical trick is used to modify the scoring function globally.

Dynamic Pharmacophore-Based Approach¶
The dynamic pharmacophore-based approach is another method that exploits the MPS information. In the first step a pharmacophore hypothesis containing a set of key features (e.g. hydrogen bond donor, acceptor, hydrophobic region, etc.) is generated using a probe for each protein of the ensemble. Then, they are all combined into one dynamic pharmacophore hypothesis (see below). The last step consists of searching in a database of ligands for molecules that fit the constructed dynamic pharmacophore.

articles
Method for Including the Dynamic Fluctuations of a Protein in Computer-Aided Drug Design Heather A. Carlson, Kevin M. Masukawa, and J. Andrew McCammon J. Phys. Chem. A 103 1999
wikipedia
# Dynamic Pharmacophore Model for HIV-1 Integrase¶
The dynamic pharmacophore-based approach was first introduced in 1999 by Carlson et al. in a study of HIV integrase. In order to validate their method, they constructed both a static and a dynamic pharmacophore model. The static model was unable to identify any of the 59 known HIV integrase inhibitors whereas most of them fitted successfully in the dynamic model. The dynamic pharmacophore model was then used to screen the ACD database, and this led to the discovery of several new inhibitors.

articles
Developing a Dynamic Pharmacophore Model for HIV-1 Integrase Carlson, H. A.; Masukawa, K. M.; Rubins, K.; Bushman, F. D.;Jorgensen, W. L. et al. J. Med. Chem. 43 2000 10.1021/jm990322h
Method for Including the Dynamic Fluctuations of a Protein in Computer-Aided Drug Design Heather A. Carlson, Kevin M. Masukawa, and J. Andrew McCammon J. Phys. Chem. A 103 1999
Dynamic Receptor-Based Pharmacophore Model Development and Its Application in Designing Novel HIV-1 Integrase Inhibitors Deng J. et al, J. Med. Chem. 48 2005
# Hybrid Pharmacophore Models using LigandScout¶
LigandScout is a recent software tool that derives pharmacophores from structure-based data. It enables the creation of hybrid pharmacophore models that can be used for virtual screening.

articles
LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters Gerhard Wolber and Thierry Langer J. Chem. Inf. Model 45 2005 10.1021/ci049885e
Domain Movements¶
Domains motions are often key determinants in the mechanism and the functions of a protein (for example the interconversion between the closed-inactive and the open-active conformations of an enzyme). Three mechanisms were identified in the movement of domains: intrinsic flexibility, hinged domain movements and ball-and-socket motions. Current effort aims at simulating such motions in computational docking by computer modeling.
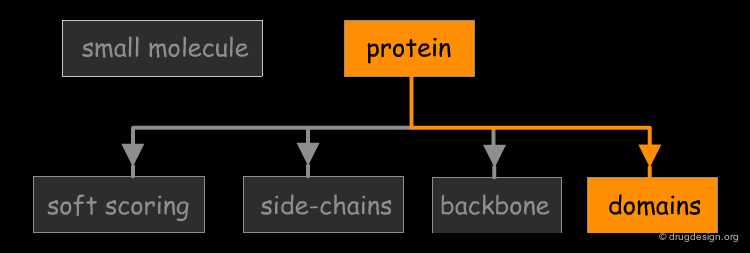

articles
Structural mechanisms for domain movements in proteins Gerstein, M.; Lesk, A.; Chothia Biochemistry 33(22) 1994
Media
This picture was made using the HingeProt server and VMD HingeProt Server
Example of Calmodulin Domain Movements¶
Studies were made to elucidate the binding of Calmodulin to a 26-residue peptide derived from a Myosin light chain kinase. Calmodulin mediates a large number of cellular functions including ion channels and protein synthesis. It is made of two domains connected by a tether. Important domain motions occur in Calmodulin upon binding. In the figure below is shown the protein before and after binding (as determined by NMR analyses).

articles
Isotope-Filtered 2D NMR of a Protein-Peptide Complex: Study of a Skeletal Muscle Myosin Light Chain Kinase Fragment Bound to Calmodulin Mitsuhiko Ikura and Ad Bax J. Am. Chem. Soc. 114 1992
Conventional Modeling Methods are not Suited¶
Conventional modeling methods for studying the dynamics of a protein are not suited to domain motions. Among these, Molecular Dynamics and Monte Carlo simulations require computational time that is far beyond practical applications for large-scale motions. New approaches have emerged that incorporate flexibility in the docking by considering those residues that play a pivotal role in these motions.

Intrinsic Flexibility¶
Intrinsic flexibility can lead some proteins to encounter large motions of domains. These can be due to small conformational changes of the backbone torsional angles that have a cumulative effect. Such motions are very difficult to simulate in computational docking.

Media
This picture was made using the HingeProt server and VMD HingeProt Server
Hinge-Bent Movements¶
Hinge bending movements are similar to rotations around an articulated joint, in that they involve a small number of residues - in some cases even one bond can provide the required rotational freedom. They are observed in many proteins such as in tobacco bushy stunt virus or in lactoferrin. Tailor-made automated algorithms have recently been developed in the modeling of hinge bending movements. Some of these are presented in the following pages.

articles
FlexOracle: predicting flexible hinges by identification of stable domains Flores SC, Gerstein MB. BMC Bioinformatics 8 2007 None
HingeProt: Automated Prediction of Hinges in Protein Structures. Emekli U, Schneidman-Duhovny D, Wolfson HJ, Nussinov R, Haliloglu T. Proteins in press 2007 None
Flexible docking allowing induced fit in proteins: insights from an open to closed conformational isomers Sandak, B.; Wolfson, H.; Nussinov, R Proteins: Structure, Function, and Genetics 32 1998 None
A Method for Biomolecular Structural Recognition and Docking Allowing Conformational Flexibility Sandak, B.; Nussinov, R.; Wolfson, H J. Comp. Biol 5(4) 1998 None
Geometry based flexible protein docking Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H. Proteins: Structure, Function, and Genetics 60 2005 None
Automated Methods for Hinge Detection¶
If a number of structures in different conformations for a protein are available, flexible protein alignment can be used to identify potential hinge locations. Another option is to use Normal Mode Analysis. Otherwise, hinges can be placed on domain linkers, loops or protein missing parts that could not be determined by X-ray crystallography.

articles
Flexible protein alignment and hinge detection. Shatsky M, Nussinov R, Wolfson HJ. Proteins 48(2) 2002 None
Incorporating Hinge-Bent Movements in Docking¶
Hinge bent movements can easily be incorporated in computer vision based docking approaches. This approach makes it possible to apply full flexible docking, efficiently explore the space of all possible conformations, and avoid the brute force search. The method can handle docking with hinge-bending movements in one of the molecules. The hinges are specified in advance; they can be placed at points and at bonds of one of the docked molecules.

Docking with Hinge-Bent Movements¶
The flexible molecule is divided into rigid parts by hinge points and all parts are docked independently. An efficient graph algorithm is then used to detect the highest scoring consistent configurations of the candidate rigid part placements. Two solutions for neighboring rigid parts are considered consistent if they superimpose the hinge point into the same position and there is no steric clash between them. The parts acquire a conformation that produces the largest shape complementarity between the molecules, resembling the induced-fit. The run-time performance of this method is comparable to the one needed for rigid docking.

Ball-and-Socket Motions¶
Ball-and-socket motions involve the rotation of a variable domain with respect to a constant one by a combination of hinge and shear motions. For example, the variable domain of an antibody can rotate by about 50 degrees with respect to its constant domains with this type of mechanism. It is still a challenge to simulate this type of flexibility in computational docking.
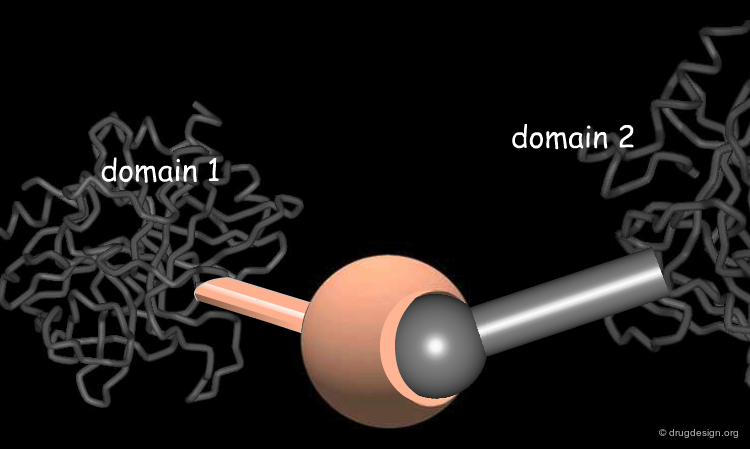
Uses of Docking in Research¶
Computational Docking in Drug Discovery¶
The ability of molecular modeling to simulate molecular recognition has proven to be of critical value in our genomic era. Docking software is used by scientists dealing with 3D mechanisms occurring in cellular events: molecular modelers and computational chemists use docking for drug discovery, biochemists to elucidate mechanisms of action, crystallographers for structure refinement, combinatorial chemists for designing new libraries etc... Docking has become a key tool in the pharmaceutical and biotech industry. In the following pages we present some typical applications of molecular docking in drug discovery.

Virtual Screening¶
Docking studies consist of predicting the 3D structure of the complex between a small molecule and a protein. When the goal is to dock all the compounds of a library (the molecules being available or not yet synthesized), the process is called virtual screening or high through-put docking. Virtual screening identifies active compounds in a large database and ranks them by their affinity to the receptor. All the examples that follow address both single and high through-put docking.

articles
Discovery of a potent and selective protein kinase CK2 inhibitor by high-throughput docking Vangrevelinghe E, Zimmermann K, Schoepfer J, Portmann R, Fabbro D, Furet P. J Med Chem 46(13) 2003
Docking and scoring in virtual screening for drug discovery: methods and applications Kitchen DB, Decornez H, Furr JR, Bajorath J. Nat Rev Drug Discov 3(11) 2004
Virtual screening and fast automated docking methods Gisbert Schneider and Hans-Joachim Bohm DDT 7(1) 2002 10.1016/S1359-6446(01)02091-8
wikipedia
Lead Hopping¶
Based on the structure of an active compound, "lead hopping" consists of the identification of novel structures with different topologies that still show the same activity. Molecular docking can be used for that purpose as described in the scheme below. The resulting molecule has a novel structure and may show increased activities, as compared to that of the initial lead.
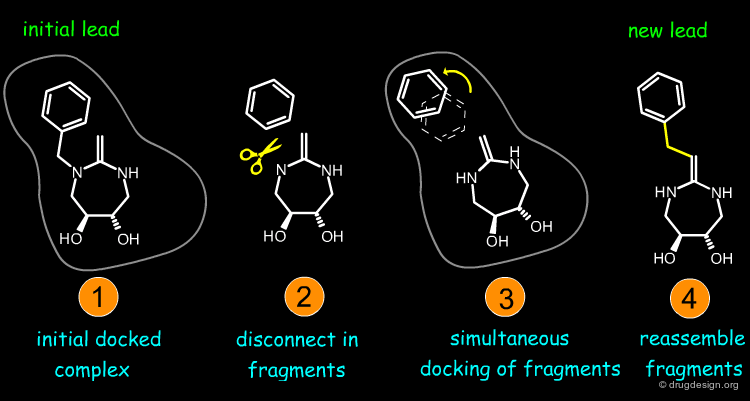
Increasing HTS Hit Rates¶
The most widely used (and also successful) type of docking today involves the selection of molecules to be processed for subsequent high through-put screening (HTS). The method is not used to recognize active molecules but to eliminate those that are likely to be inactive.
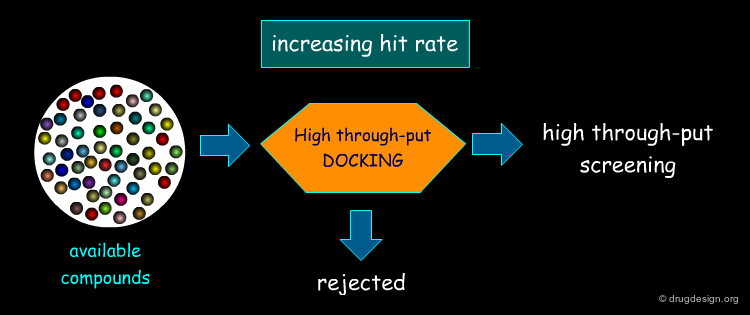
wikipedia
Confirm Choice of Prototype Structure¶
When embarking on a new project when the competition is already active, the choice of the starting point is of great importance. When a promising scaffold is identified, docking experiments are an excellent move to assess the consequences of this choice. A good grasp of the interactions with the protein can lead to more creative and a better scaffold. Reducing the risks of failure increases the chances of success. In the example below, a scaffold was selected based on 3D-QSAR analyses; docking treatments were carried out to get a 'second opinion' to confirm the validity of this selection.

Manual Design of a New Scaffold¶
The information about a docked complex can be directly exploited in modifying the structure of the ligand and designing a new one, using manual interactive modeling methods. In the example below, an initial docked model (ligand in red) was the basis for the design of a novel scaffold (in blue) with improved anchorage capabilities.
New Cores from a Database of Scaffolds¶
Starting from a docked complex, and based on databases of scaffolds, high through-put docking can be done to reveal candidate structures that exploit the same or more interactions with the protein.
De Novo Design of Spacers¶
The information from a docked complex can be directly exploited by keeping the fragments interacting directly with the protein, and removing the spacer or the scaffold that holds them. Automated de novo design programs can be then used along with libraries of fragments to find new spacers that position the interacting fragments correctly in 3D.
Modulating Protein-Protein Interactions¶
In a biological system involving the interaction of two proteins, it may be therapeutically important to understand how the two proteins interact in 3D for the design of molecules blocking or activating the process. In the absence of X-ray crystals of the complex, this issue can be addressed either by docking studies or experimentally, using a single or a double point mutation approach. Docking studies help to identify residues involved in the interaction and also show which residues should be mutated. This information can be further exploited to design ligands (peptides or non-peptides) that interfere with the binding of the two proteins.

wikipedia
Query for 3D Database Searching¶
The constraints describing a query to be used for 3D database searching can be derived from docking analyses. The query is designed to search for different cores that produce the same type of interactions.

Creative Molecular Design Conditions¶
Docking can be the basis for creative molecular design, as evidenced by elegant and successful ones produced in many discovery projects. For example in a protein kinase inhibition project, the docking results of a reference inhibitor (green) superimposed on the experimental structure of the ATP cofactor (yellow) led to the design and validation by docking calculations of an entirely novel molecule (pink) that was subsequently found to be very potent and selective.
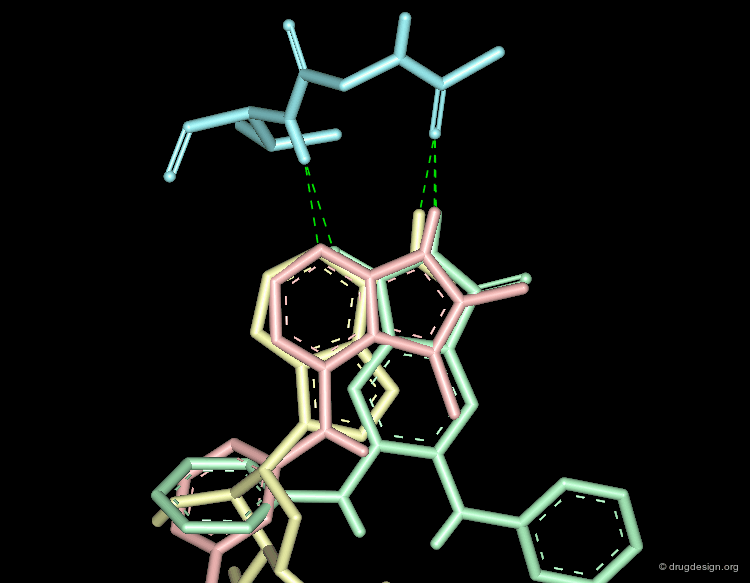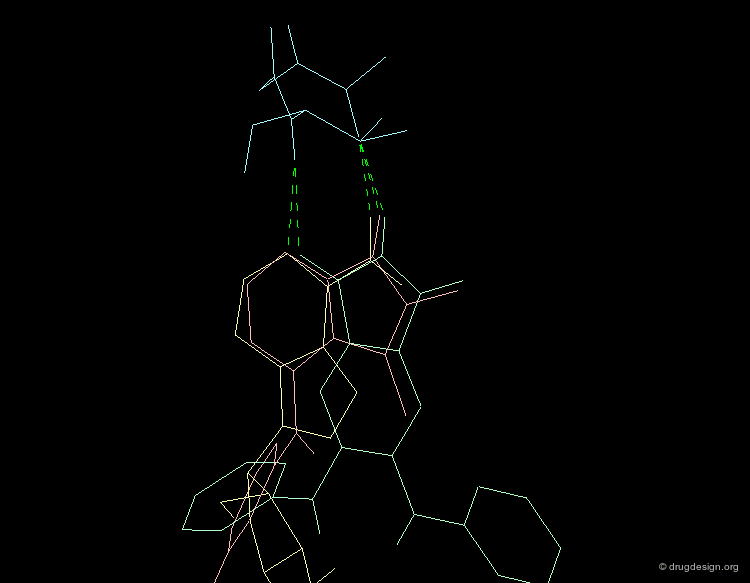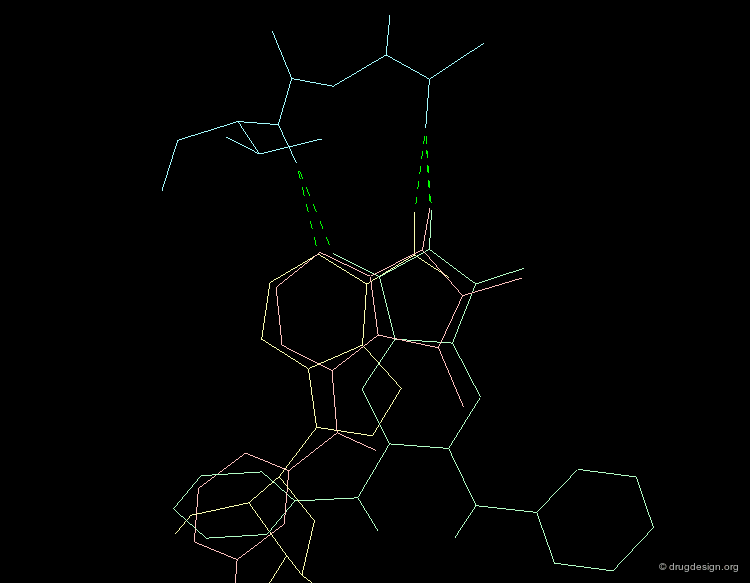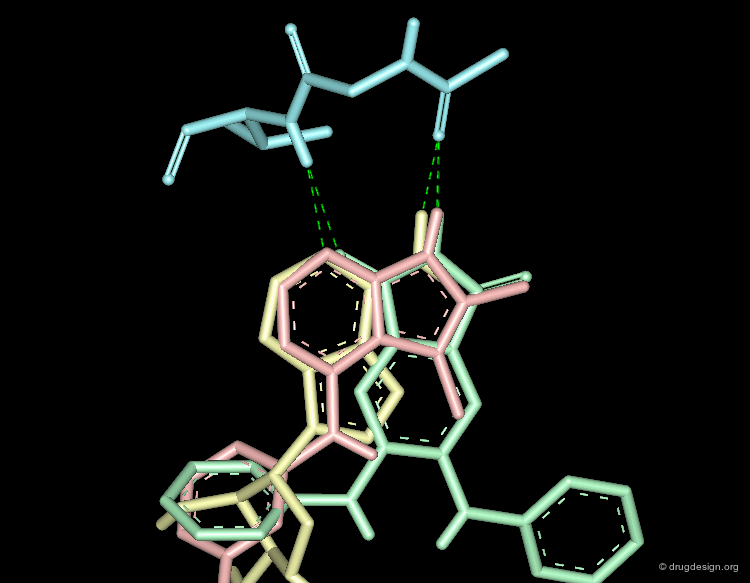
articles
Modelling Study of Protein Kinase Inhibitors: Binding Mode of Staurosporine and Origin of the Selectivity of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U, Traxler P J. Comput. Aided Mol. Des. 9 1995
4-(Phenylamino)Pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T, and Lydon N J. Med. Chem. 39 1996
Design of Combinatorial Libraries¶
Docking analyses can help reduce a huge virtual library to a practical size for combinatorial synthesis and high through-put screening. Molecules that create clashes with the receptor are immediately eliminated. Docking calculations can sort the molecules in terms of their binding capabilities and identify the best set of substituents.
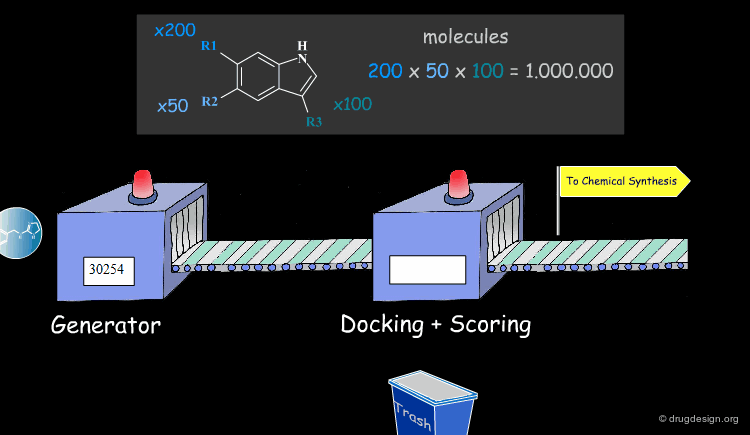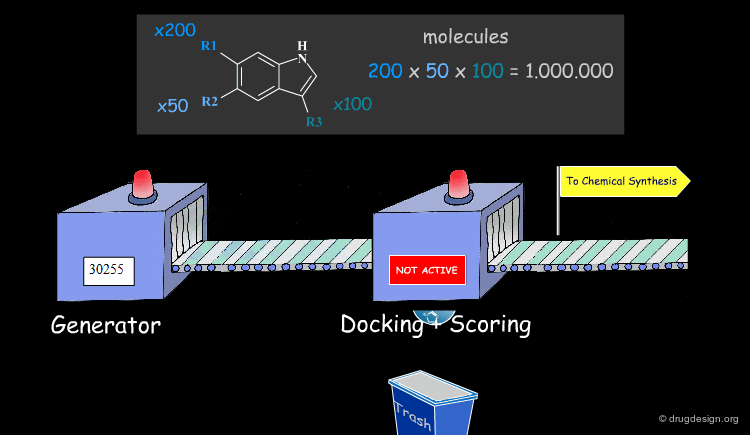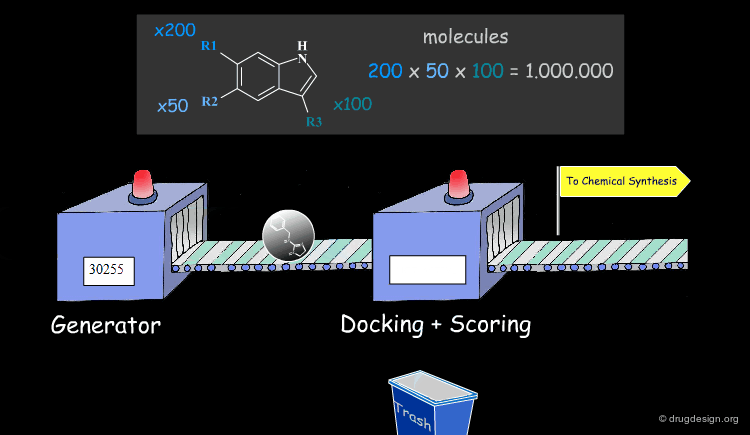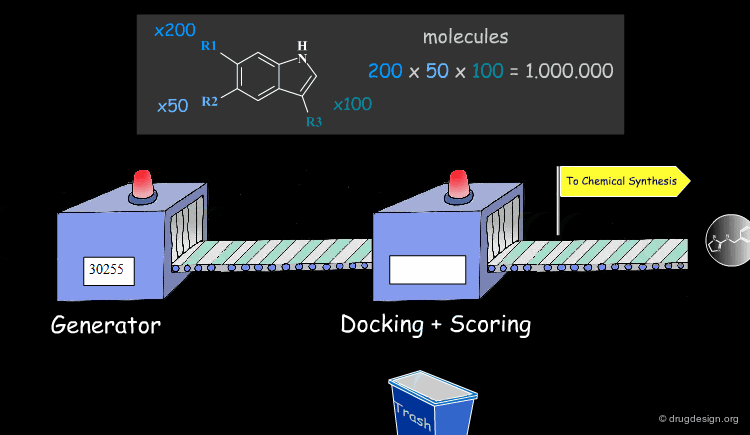
wikipedia
Understanding SAR¶
The results of docking calculations can be directly exploited to understand the structure-activity relationships (SAR) observed in a series of compounds. (Click on the biological activities indicated below to visualize and analyze the corresponding docked complex).

wikipedia
Reducing Multiple Hypotheses to a Single One¶
When assessing the binding mode of a new molecule several possibilities can sometimes be entertained; this can be further clarified with docking calculations. In the example below the molecule in question (a Cdk2 inhibitor) demonstrates that solution 1 is by far the most preferable out of the four possible anchorages. For clarity, only the anchorage with the protein is shown, whereas more residues are visualized for the preferred binding mode (button 'Preferred').

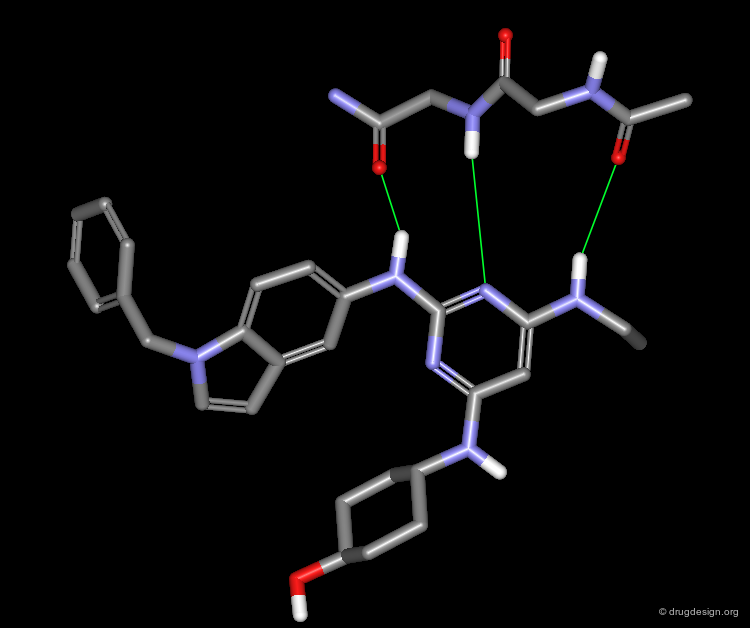




articles
Assessment of multiple binding modes in ligand-protein docking Kallblad P, Mancera RL, Todorov NP. J Med Chem 47(13) 2004
The Effect of Multiple Binding Modes on Empirical Modeling of Ligand Docking to Proteins Brem R and Dill KA Protein Sci. 8 1999
Selective In Vivo and In Vitro Effects of a Small Molecule Inhibitor of Cyclin-Dependent Kinase 4 Rajeev Soni, Terence O'Reilly, Pascal Furet, Lionel Muller, Christine Stephan, Sabine Zumstein-Mecker, Heinz Fretz, Doriano Fabbro, Bhabatosh Chaudhuri Journal of the National Cancer Institute 93(6) 2001
Series Optimization¶
The docking results of a candidate prototype compound can guide the optimization of the initial structure. Unfavorable interactions can be suppressed and additional favorable interactions can be created in the modified ligand. The significance of novel candidate molecules is then assessed with novel docking calculations.
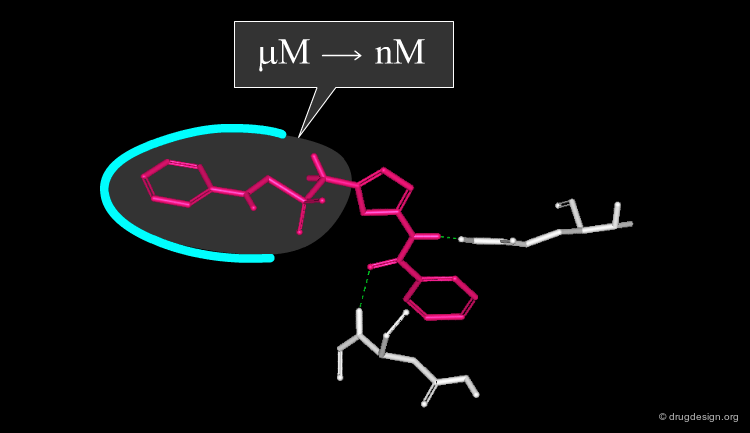
articles
2-Amino-3,4-dihydroquinazolines as Inhibitors of BACE-1 (beta-Site APP Cleaving Enzyme): Use of Structure Based Design to Convert a Micromolar Hit into a Nanomolar Lead Ellen W. Baxter, Kelly A. Conway, Ludo Kennis,Francois Bischoff, Marc H. Mercken, Hans L. De Winter, Charles H. Reynolds, Brett A. Tounge, Chi Luo, Malcolm K. Scott, Yifang Huang, Mirielle Braeken, Serge M. A. Pieters, Didier J. C. Berthelot, Stefan Masure, Wouter D. Bruinzeel, Alfonzo D. Jordan, Michael H. Parker, Robert E. Boyd, Junya Qu, Richard S. Alexander, Douglas E. Brenneman, Allen B. Reitz J.Med.Chem. 50 (18) 2007 10.1021/jm0705408
Explaining Incomprehensible Observations¶
Molecular docking is a tool of choice to help solve incomprehensible observations where small structural modifications of a ligand result in huge differences in biological activities. In the example below, the simple move of a nitrogen atom in the heterocyle transforms an inactive compound into a potent inhibitor. Docking calculations provide a reasonable interpretation for this observation (see button "Solution").

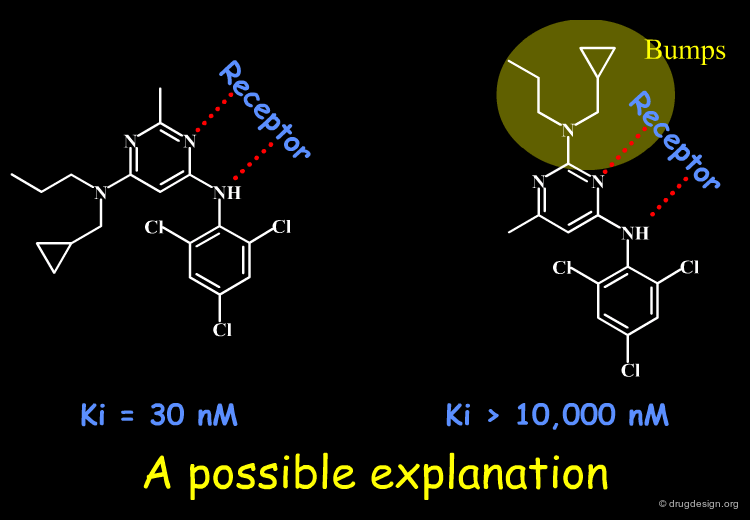
articles
Design and Synthesis of a Series of Non-Peptide High-Affinity Human Corticotropin-Releasing Factor1 Receptor Antagonists Chen Chen, Raymond Dagnino, Jr., Errol B. De Souza, Dimitri E. Grigoriadis, Charles Q. Huang, Kyung-Il Kim, Zhengyu Liu, Terry Moran, Thomas R. Webb, Jeffrey P. Whitten, Yun Feng Xie, and James R. McCarthy J. Med. Chem. 39 1996 10.1021/jm960149e
book
Wermuth CG Rational Approaches to Drug Design Prous Science 2001
Identifying Incorrect Working Hypotheses¶
Adenine-based libraries were synthesized as probes for cellular processes involving ATP. Many X-ray studies showed that ATP and adenine-like analogs are bound in the same orientation, and this was thought to be general. X-ray studies however found Olomoucine in a different orientation, invalidating the hypothesis and docking studies were able to fully reproduce this observation. This and other predictions based on docking analyses presented in other sections of this course, demonstrate that docking is well suited for the identification of incorrect working hypotheses.


articles
Multiple Modes of Ligand Recognition: Crystal Structures of Cyclin-dependent Protein Kinase 2 in Complex with ATP and Two Inhibitors, Olomoucine and Isopentenyladenine Schulze-Gahmen U, Brandsen J, Jones HD, Morgan DO, Meijer L, Vesely J and Kim SH Proteins 22 1995
Crystal Structure of Cyclin-Dependent Kinase 2 De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO and Kim SH Nature 363 1993
Inhibition of Cyclin-dependent Kinases by Purine Analogues Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S and Meijer L Eur. J. Biochem. 224 1994
Structural basis of inhibitor selectivity in MAP kinases Wang Z, Canagarajah BJ, Boehm JC, Kassisa S, Cobb MH, Young PR, Abdel-Meguid S, Adams JL, Goldsmith EJ Structure 6 1998
Align Chemically Unrelated Molecules in 3D¶
It is extremely difficult to superimpose chemically unrelated active compounds that are known to bind to the same receptor site. Not only are their bioactive conformations not known but also it is not clear which fragments must be aligned. Molecular docking provides powerful means to reveal their probable bioactive conformation, their alignment and the 3D relationships between the different moieties of each molecule. This is useful for many purposes including hybrid design and aligning molecules for 3D-QSAR studies.

articles
Optimization of a pharmacophore model for 5-HT4 agonists using CoMFA and receptor based alignment Magdy N. Iskander, Lok M. Leung, Trevor Buley, Fadi Ayad, Juliana Di Iulio, Yean Y. Tan and Ian M. Coupar European Journal of Medicinal Chemistry 41 (1) 2006
A CoMFA study of COX-2 inhibitors with receptor based alignment Prasanna A. Datar, Evans C. Coutinho Journal of Molecular Graphics and Modelling 23 2004
Three-dimensional quantitative structure-activity and structure-selectivity relationships of dihydrofolate reductase inhibitors Jeffrey J. Sutherland and Donald F. Weaver Journal of Computer-Aided Molecular Design 18 2004
3D-QSAR CoMFA, CoMSIA studies on substituted ureas as Raf-1 kinase inhibitors and its confirmation with structure-based studies Ram Thaimattam, Pankaj Daga, Shaikh Abdul Rajjak, Rahul Banerjee and Javed Iqbal Bioorg Med Chem 12 2004
Improving the Solubility of a Ligand¶
The docking of a potent inhibitor but a poorly soluble one can be exploited to introduce an additional group designed to enhance its solubility. In the example below (EGF-R inhibitors) a quinazoline structure with two methoxy substituents was discovered with an IC50 of 29 picomolar; however it is highly insoluble. One of the methoxy groups was then extended with a terminal morpholino group (shown in the view). The design aimed not to interfere with the protein and allowed the morpholino group to reach the solvent area outside the protein. This design was a success and the molecule was further developed and commercialized (Iressa).

articles
Chemical Inhibitors of Protein Kinases Bridges AJ Chem. Rev 101 2001
Understand the Intrinsic Limitations of a Scaffold¶
Docking calculations can reveal the intrinsic limitations of a scaffold. For example, the unsubstituted dioxobenzothiazole molecule shown below was found to inhibit Cdk4 with an IC50 of 6 µM. Optimization of the series failed to find more potent compounds: docking analyses reveal that for this scaffold, there are undesirable steric and polar repulsions between the nitrogen atom of the thiazole and the carbonyl of the hinge, a characteristic found in all analogs synthesized.
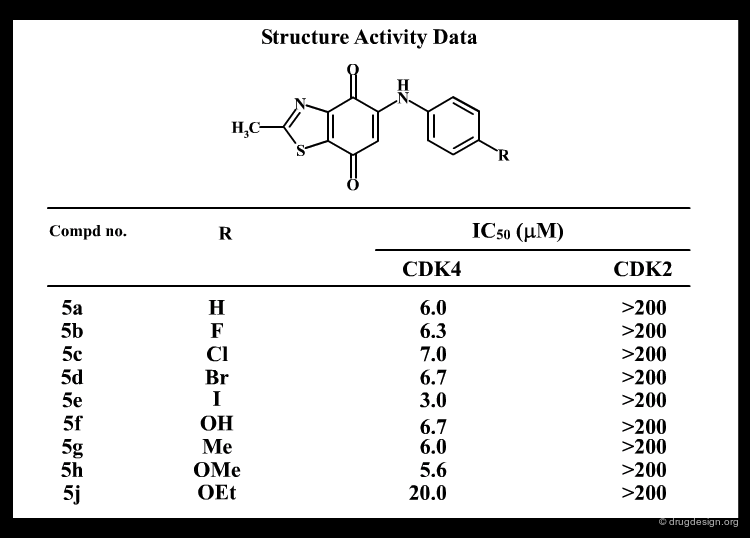

articles
5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents Ryu CK, Kang HY, Lee SK, Nam KA, Hong CY, Ko WG and Lee BH Bioorg. Med. Chem. Lett 10 2000
Assessing the Potential of a Hit¶
When a hit is discovered by high through-put screening, it might not be structurally suited for optimization. Perhaps the structure does not permit access to favorable interactions with the receptor? Perhaps it contains some detrimental interactions? Perhaps a nitrogen atom in a heterocycle should be replaced by a carbon to enable the introduction of substituents? Answers to these questions can be addressed by docking calculations that provide precious information on the potential of the hit and desirable modifications.
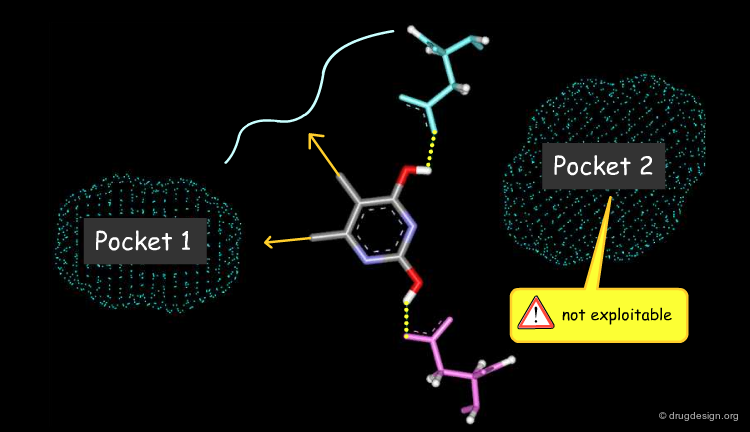
Elucidating Exact Mode of Action¶
Docking studies can help elucidate the exact mode of action of a system. For example thymidylate synthase can be inhibited by ligands binding either to the substrate site or to the cofactor site. In the example below, the inhibitor (in yellow) does not compete with the cofactor (in blue): it occupies another region of the catalytic site and at the same time establishes favorable interactions with the cofactor. Computational docking is a good alternative to experimental studies.

articles
Design of Thymidylate Synthase Inhibitors Using Protein Crystal Structures: the Synthesis and Biological Evaluation of a Novel Class of 5-Substituted Quinazolinones Webber SE, Bleckman T, Attard J, Deal JG, Katherdekar V, Welsh KM, Webber S, Janson CA, Matthews, DA, Smith WW, Freer ST, Jordan SR, Bacquet RJ, Howland EF, Booth CJL, Ward RW, Hermann SM, White J, Morse CA, Hilliard JA and Bartlett CA J. Med. Chem. 36 1993
Thymidylate Synthase Inhibition: A Structure-Based Rationale for Drug Design Costi MP Med. Res. Rev. 18 1998
Structure-Based Design of Lipophilic Quinazoline Inhibitors of Thymidylate Synthase Jones TR, Varney MD, Webber SE, Lewis KK, Marzoni GP, Palmer CL, Kathardekar V, Welsh KM, Webber S, Matthews DA, Appelt K, Smith WW, Janson CA, Villafranca JE, Bacquet RJ, Howland EF, Booth CL, Herrmann SM, Ward RW, White J, Moomaw EW, Bartlett CA and Morse CA J. Med. Chem. 39 1996
Assessing Multiple Alignment Hypotheses¶
When comparing chemically unrelated active molecules in a pharmacophore-based perspective, it is possible to generate many possible alignments of the molecules; however, which is the correct one? Docking calculations can help resolve different hypotheses and discriminate between them.
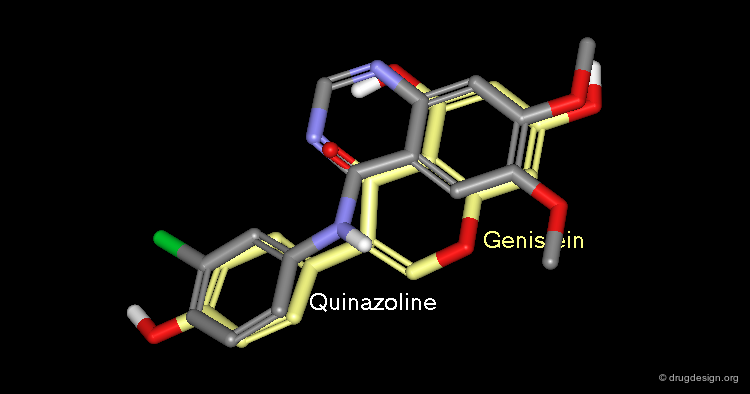




Molecular Mimicry¶
Molecular docking calculations are useful for predicting the bioactive conformation of a ligand. This information can be further exploited for molecular mimicry purposes, based for example on the superimposition with the bioactive conformations of other active molecules, in a pharmacophore-based design approach.
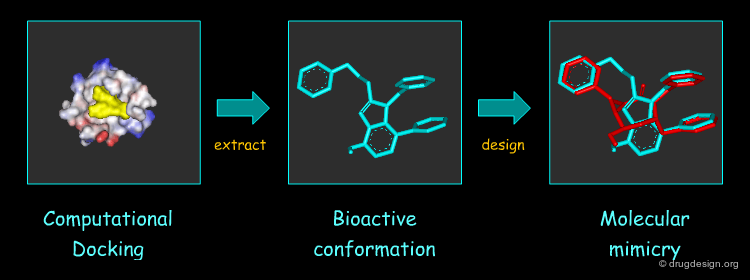
Computational Validation of Hypotheses¶
Docking can be used to validate a hypothesis. In the example here the Z stereochemistry was suspected to be the bioactive geometry of methyl-idenyl-indolin-2-one derivatives. This hypothesis can be tested either computationally (with docking calculations) or experimentally (by making a molecule with a preferred Z geometry). The response time to such questions is much faster with computational studies than through the synthesis of new molecules.

articles
Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases Sun L, Tran N, Tang F, App H, Hirth P, McMahon G and Tang C J. Med. Chem 41 1998
Docking Softwares¶
Docking Programs¶
Docking programs started to appear in the late 1980s. DOCK was the first one to be introduced. Current developments allow for ligand flexibility. Some programs have been developed for simulating protein-protein docking, whereas others for high throughput screening using large databases. A brief outline of some of these programs can be found in the following pages.
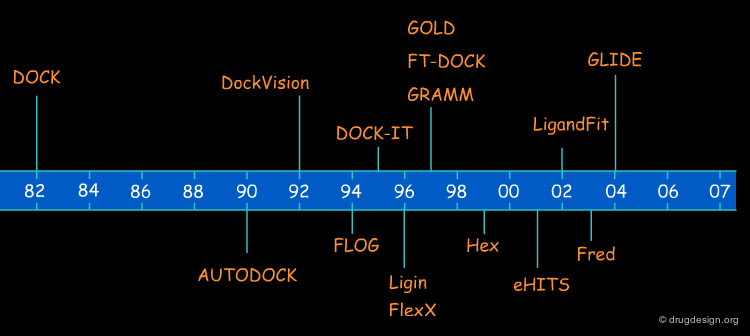
Dock¶
DOCK was the first docking program. It was introduced by Kuntz's group at UCSF. Historically, the DOCK algorithm addressed rigid body docking using a geometric matching algorithm to superimpose the ligand onto a negative image of the binding pocket. Important features that improved the algorithm's ability to find the lowest-energy binding mode, including force-field based scoring, on-the-fly optimization, an improved matching algorithm for rigid body docking and an algorithm for flexible ligand docking, have been added over the years.

articles
A geometric approach to macromolecule-ligand interactions Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 161 1982
Autodock¶
Autodock was the first program that allows docking a flexible ligand. AutoDock is a suite of automated docking tools. It is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure.

Media
AutoDock docking of camphor (white and red) to cytochrome P450cam from Pseudomonas putida with the crystal structure (green)
DockVision¶
DockVision is a docking package providing access to multiple algorithms including Monte Carlo, Genetic Algorithm, and database screening docking algorithms. The program is associated with a 3D viewer called DockCam. Below is shown a result obtained with DockVision in the docking of pentamide in the active site of trypsin.

DockIt¶
DockIt ® is well adapted to parallel computations and uses a distance geometry based approach to docking. DockIt only requires 2D connectivity information about the ligands, so it is quite easy to output some connection table type information (Daylight SMILES or MDL mol files) from a database or a combinatorial design computation through DockIt. The distance geometry engine first generates a bound conformation of the ligand which fits into the shape of the binding pocket and then refines both shape and chemical complementarity. This is different from many other docking programs which first place a few electrostatic interactions (hydrogen bonds typically) and then refine the shape complementarity. Thus, DockIt is complementary to most other docking programs.

FlexX¶
FlexX docks a conformationally flexible ligand into a binding site, using an incremental construction algorithm that builds the ligand in the site. The resulting docked conformations are scored based on the strength of ligand-receptor interactions. Receptor flexibility, pharmacophore constraints and other functionalities can be taken into account using special add-on modules. FlexX holds bond lengths and angles invariant, using the values of the input ligand. Each acyclic single bond is assigned up to 12 dihedral angle values - values based on a statistical evaluation of fragments in the Cambridge Structural Database.

Ligin¶
LIGIN, developed by V. Sobolev et al., is publicly available to academic institutions. The method is based on surface complementarity between ligand and receptor, taking into account the shape and the chemical nature of the atoms. The program has been further developed to incorporate ligand flexibility. This software has been tested during CASP2 experiments to predict structures of seven ligand-protein complexes. In six cases the location of the binding pocket was identified correctly (one of them is shown below); in two cases the ligand was docked correctly; in three cases the exposed part of the ligand was docked poorly, although the buried parts were docked well.

FT-Dock¶
FT-Dock belongs to a suite of programs designed to enable computational prediction of protein-protein docking. The original version was written in 1997 by Henry Gabb with parallel capabilities for a Silicon Graphics Challenge machine. The current version was written by Gidon Moont with no parallel capabilities but is portable to many platforms. The FT-Dock algorithm is based on the Katchalski-Katzir (a Fourier Transform rigid body docking). It discretizes the two molecules onto orthogonal grids and performs a global scan of translational and rotational space. The scoring method is a surface complementarity score between the two grids.
articles
Modelling Protein Docking using Shape Complimentarity, Electrostatics and Biochemical Information Henry A. Gabb, Richard M. Jackson, Michael J. E. Sternberg Journal of Molecular Biology 272 1997 10.1006/jmbi.1997.1203
Use of pair potentials across protein interfaces in screening predicted docked complexes. G. Moont, H. A. Gabb, and M. J. E. Sternberg. Proteins 35(3) 1999
Rapid refinement of protein interfaces incorporating solvation: application to the docking problem. R. M. Jackson, H. A. Gabb, and M. J. E. Sternberg J. Mol. Biol., 276(1) 1998
Modelling Protein Docking using Shape Complimentarity, Electrostatics and Biochemical Information Patrick Aloy, Gidon Moont, Henry A. Gabb, Enrique Querol, Francesc X. Aviles, Michael J. E. Sternberg Proteins: Structure, Function, and Genetics 33 1998
GOLD¶
GOLD is a program for calculating the docking modes of small molecules into protein binding sites. GOLD features include a genetic algorithm for protein-ligand docking, full ligand and partial protein flexibility, energy functions partly based on conformational and non-bonded contact information from the CSD, choice of scoring functions (GoldScore, ChemScore and now user-defined score). Below is illustrated the result obtained by docking the ligand shown in 1JAP, a matrix metalloprotease. The GOLD docked solution (in red) is compared with the solution obtained experimentally (colored by element).
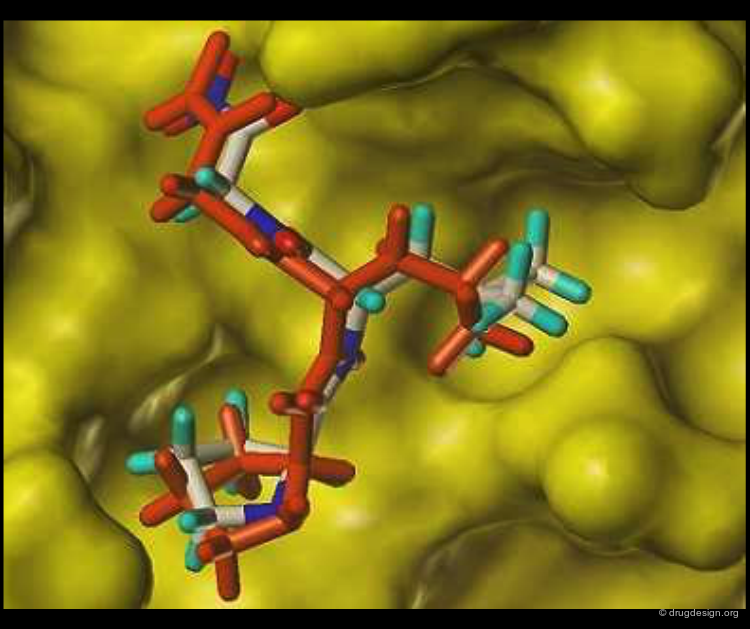
GRAMM¶
GRAMM is a free FFT-based docking program. The molecular pairs can be two proteins, a protein and a smaller compound, two transmembrane helices, etc. To predict the structure of a complex, it only requires the atomic coordinates of the two molecules (no information about the binding sites is needed). The program performs an exhaustive 6-dimensional search through the relative translations and rotations of the molecules. A web-version is available for low-resolution docking treatments.

Hex¶
Hex is an interactive protein docking and molecular superposition program written by Dave Ritchie at the University of Aberdeen. Hex differs primarily from other macromolecular docking programs and molecular graphics packages in its use of polar Fourier correlations to accelerate docking calculations. The graphical nature of Hex came about largely because the author wanted to visualize the results of such docking calculations in a natural and seamless way, without having to export unmanageably many (and usually quite big) coordinate files to one of the many existing molecular graphics packages.

eHiTS¶
eHiTS is a docking program for virtual high throughput screening of large databases. The program has a scoring function that takes advantage of temperature factor information provided in PDB files to give a more complete picture of the interactions. The program also uses the probability of the atom position to create a statistically derived empirical scoring function.

articles
eHiTS: A new fast, exhaustive flexible ligand docking system Zsolt Zsoldos, Darryl Reid, Aniko Simon, Sayyed Bashir Sadjad, A. Peter Johnson Journal of Molecular Graphics and Modelling 26 2007
LigandFit¶
In LigandFit the active site is identified using a flood filling technique. Docking is conducted with constraint filters (H-bond donor, H-bond acceptor, hydrophobic contact, and metal ion bond) and user-defined receptor interactions. The program contains several scoring functions including LigScore, PLP14, PLP25, and PMF6. In addition, consensus scoring can also be performed using a combination of functions.

FRED¶
FRED is a protein-ligand docking program that receives as an input a multiconformer database of one or more small molecules, a target protein structure, a box defining the active site of the protein and several optional command-line parameters. The ligand conformers and protein structure are treated as rigid during most of the docking process. The search is based on solid-body rotations and translations of each conformer. Below is shown a result obtained for the Napap inhibitor (magenta) in the active site of human thrombin (green).

Glide¶
Commercialized by Schrodinger L.L.C. Glide is a docking program suited for high-throughput virtual screening. A systematic search of the conformational, orientational, and positional space of the docked ligand is performed. Millions of compounds can be screened in a reasonable amount of time with this program.
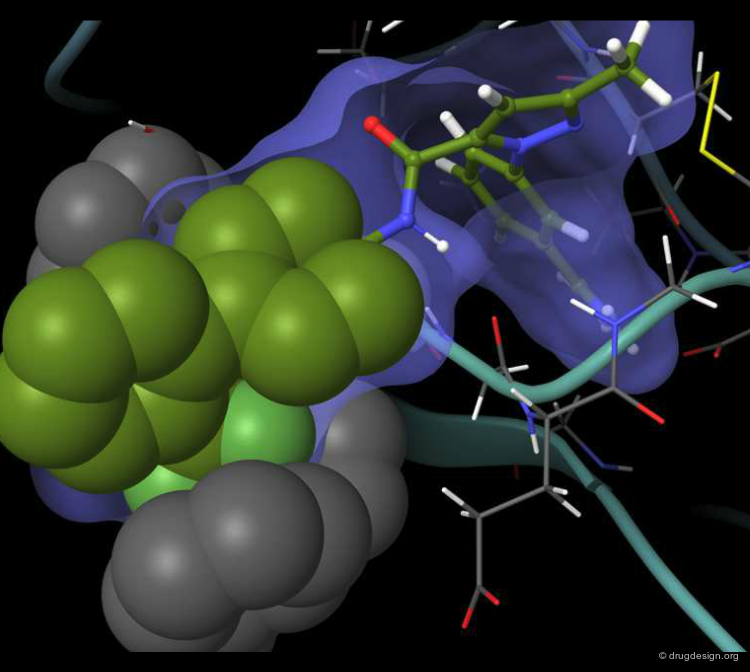
articles
Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy Richard A. Friesner, Jay L. Banks, Robert B. Murphy, Thomas A. Halgren, Jasna J. Klicic, Daniel T. Mainz, Matthew P. Repasky, Eric H. Knoll, Mee Shelley, Jason K. Perry, David E. Shaw, Perry Francis, and Peter S. Shenkin J. Med. Chem. 47 (7) 2004
Media
A top-ranking Factor Xa ligand docked by Glide using XP mode. Ligand atoms within a region of hydrophobic enclosure that strongly contribute to enhanced binding affinity are illustrated in CPK.
Which Software is Better?¶
Most experts agree that no current tool has consistently demonstrated superior performance across a broad range of systems. Researchers in the pharmaceutical industry carry out comparative studies of docking and scoring methods on their own targets, to identify which method (or combination of methods) works best. The diagram below lists the most popular software cited in the literature in 2005.

articles
Protein-ligand docking: Current status and future challenges Sousa S.F.
10.1002/prot.21082
Comparing protein-ligand docking programs is difficult Cole JC, Murray CW, Nissink JW, Taylor RD, Taylor R. Proteins 60(3) 2005 10.1002/prot.20497
Future and Perspectives¶
Limitations in Computational Docking¶
Computational docking has emerged recently as a new discipline. Despite the important achievements that have been obtained, substantial progress remains to be made to exploit the full potential of this approach. Current limitations, key issues and future perspectives are presented in the pages that follow.

Trade Off Between Efficiency and Accuracy¶
Molecular docking is above all a computational challenge that involves juggling the exigencies of efficiency on the one hand and accuracy on the other. Knowing that approximations inevitably introduce some loss in accuracy the question is how to obtain correct solutions without increasing substantially the computational time. The efficiency/accuracy tradeoff is an issue of primary importance in the development of new methods in computational docking.

Screening Large Chemical Libraries¶
One outcome of the tradeoff between efficiency and accuracy is that current computational docking software cannot be used to screen a large number of molecules in a reasonable amount of time. This key problem is magnified considerably when the virtual screening address several biological targets in parallel (e.g. for selectivity).

wikipedia
A Two Step Strategy¶
To increase accuracy while restraining computational costs, some groups use a strategy in which a two-step process is applied. The first step is to use a speedy low resolution method which filters out bad candidates; then a high resolution and more computationally intensive method is applied to obtain more accurate predictions. An example of such an algorithm is presented here.

articles
Protein-Protein Docking with Simultaneous Optimization of Rigid-body Displacement and Side-chain Conformations Gray. J. J. et al. J. Mol. Biol 331 2003 10.1016/S0022-2836(03)00670-3
High-throughput Docking Using Grid-Computing¶
Another strategy to tackle the accuracy/efficiency issue is to increase computational power. Grid computing has recently emerged for solving massive computational problems by exploiting the unused resources of disparate computers treated as a virtual cluster. It was successfully exploited for high-throughput docking in areas including cancer, AIDS and malaria.

articles
The Virtual Laboratory: a toolset to enable distributed molecular modelling for drug design on the World-Wide Grid Buyya R. et al. Concurrency and Computation: Practice and Experience (CCPE) Journal 15(1) 2003 10.1002/cpe.704
The Virtual Laboratory: a toolset to enable distributed molecular modelling for drug design on the World-Wide Grid Buyya R, Branson K, Giddy J, Abramson D Concurrency Computat.: Pract. Exper. 15 2003 10.1002/cpe.704
wikipedia
How Does it Work?¶
The diagram shown below summarizes the typical steps involved in Grid Computing docking. The user starts by downloading and installing a small application, the program will download the target and the first candidate molecule from the server. The software will generate conformers: each one is docked independently. The best solutions are returned to the server and a new candidate compound is downloaded. The process is then repeated with a new molecule. It is important to note that the docking runs during idle time where the computer is not in use.
Wide In Silico Docking On Malaria (WISDOM)¶
An example of successful implementation of grid computing applied to docking is the WISDOM project. The experiment took place between October 2006 and January 2007 using a computing Grid spanning 27 countries. Over 140 million compounds were docked against target proteins of the malaria parasite. The project used the equivalent of 420 years of computing power from a single PC with up to 5000 computers being used simultaneously. The best molecules are now being tested biologically. This project demonstrated that the difficult task of doing large-scale computational docking can be turned into a feasible one.

wikipedia
Enrichment Factor¶
An important measure for assessing the performance of a docking experiment is the enrichment factor. This is defined as the ratio between the percent of active molecules in the sample portion and the percent of active molecules in the entire database. For example, let us consider a database of 200,000 molecules containing 200 active molecules. A random selection gives a hit-rate of 0.1%. A docking program may lead to the selection of 1,000 hits. If the biological tests reveal that 30 molecules out of the 1,000 correspond to active compounds, we would have an enrichment factor of 30-fold over the random selection. A good docking algorithm should give high values for the enrichment factor for a broad range of receptor types.


Current Status of the Docking Problem¶
A recent study done by the Shoichet group provides a good reflection of current docking predictions. A docking study was done on 10 different enzymes using their holo, apo, and homology modeled structures with a database of small ligand molecules. The enrichment was found for the holo, apo and modeled to be 70%, 20% and 10%, respectively. These results suggest that substantial progress remains to be made to address real docking problems. On the next page bottlenecks associated with these results are reviewed.

articles
Information Decay in Molecular Docking Screens against Holo, Apo, and Modeled Conformations of Enzymes McGovern SL, Shoichet BK J Med Chem 46(14) 2003 10.1021/jm0300330
The Docking Bottlenecks¶
Although many bottlenecks exist, the most critical are: scoring, flexibility and protein modeling. Bound docking is limited by scoring; unbound docking by scoring and flexibility; and modeled design is limited by all three. The bottleneck in protein design is beyond the scope of the present chapter and will be presented elsewhere. Strategies to develop more effective scoring functions and better incorporation of flexibility are presented on the following pages.

More Effective Scoring Functions¶
The development of effective scoring functions is a central issue in computational docking and substantial effort is still needed for the effective scoring of solvation and entropy factors. A docking program that incorrectly discriminates between good and bad ligands or gives incorrect bound conformations is problematic. In the meantime users must use imperfect scoring functions hoping that good molecules will be placed in top positions. Moreover, because of the complexity of binding, correct docking and a good scoring function do not always mean correct binding energy.

articles
Scoring functions for protein-ligand interactions: a critical perspective Tanja Schulz-Gasch, Martin Stahl Drug Discovery Today 1(3) 2004 10.1016/j.ddtec.2004.08.004
Recent Advances in Docking and Scoring E. M. Krovat, T. Steindl and T. Langer Current Computer-Aided Drug Design 1 2005
Modeling the Solvent¶
Solvent effects are complex and are often ignored in computational docking. Simplified solvent models have been identified to be a major cause of inaccuracy in docking results. The most rigorous methods consider explicit solvent molecules but they are too slow for database screening. Other approaches represent the solvent as a continuum (for example using a fixed dielectric constant) or calculate binding energies without solvent, and then correct the values by the solvation energies calculated for the isolated ligands.

Validation of Scoring Functions¶
The availability of databases containing protein-ligand complexes with their experimental binding affinity is of paramount importance for the development and refinement of good scoring functions. The PDBbind database developed by the Wang group at the University of Michigan provides a collection of experimentally measured binding affinity data (Kd, Ki, and IC50) of 900 protein-ligand complexes available in the Protein Data Bank (PDB).

articles
The PDBbind Database: Methodologies and updates Wang, R.; Fang, X.; Lu, Y.; Yang, C.-Y.; Wang, S. J. Med. Chem. 48(12) 2005
The PDBbind Database: Collection of Binding Affinities for Protein-Ligand Complexes with Known Three-Dimensional Structures Wang, R.; Fang, X.; Lu, Y.; Wang, S. J. Med. Chem. 47(12) 2004
Target Trainable Scoring Functions¶
An all-purpose scoring function cannot treat all types of proteins equally well. A new trend in scoring function is the development of target-directed scoring. For example Hu et al. developed a tailor-made scoring function for matrix metalloproteinase derived from 40 metalloproteinases using multivariate statistical analysis. They improve the accuracy and enrichments results for the target considered.
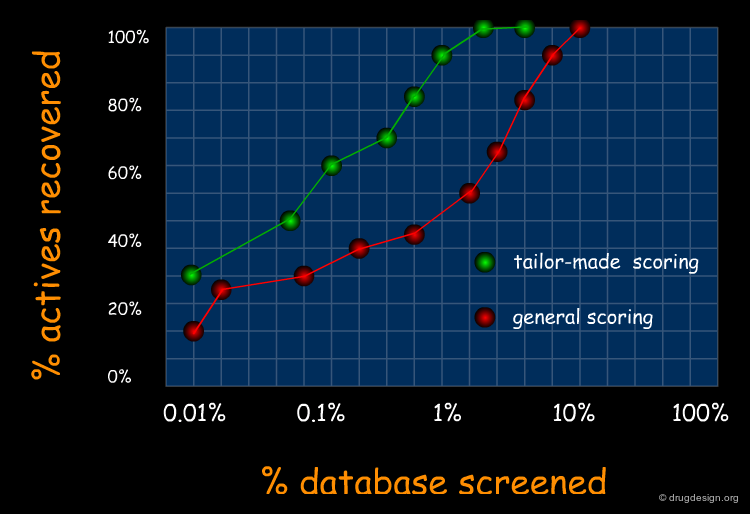
articles
Recent Advances in Docking and Scoring E. M. Krovat, T. Steindl and T. Langer Current Computer-Aided Drug Design 1 2005
book
Hu, X et al. 225th ACS National Meeting; ACS, Washington, D. C: New Orleans, LA, United States ACS 2003
Database of Decoys¶
In order to complement the tools available for the development of scoring algorithms, the UCSF has recently created a database of 3,000 compounds active against a total of 40 target proteins. For each active compound, a set of 36 "decoys" consisting of inactive molecules with similar physical properties is included in the database. The authors suggest using this benchmark for fine-tuning the scoring methods to identify good hits and eliminate false-positive hits.

articles
Benchmarking Sets for Molecular Docking Huang N., Shoichet B.K., Irwin J.J. J. Med. Chem. 49(23) 2006 10.1021/jm0608356
Consensus Scoring¶
Another strategy to improve scoring methods is to combine results from different scoring approaches. This approach is called consensus scoring, and has been successfully used to reduce the false-positive rate. Note that consensus scoring should be used with unrelated scoring functions, otherwise this could amplify calculation errors, rather than balance them.

articles
Consensus scoring for protein-ligand interactions Miklos Feher Drug Discovery Today 11 2006 10.1016/j.drudis.2006.03.009
The Molecular Flexibility Challenge¶
The second challenge in molecular docking is accounting for molecular flexibility. Considering ligand flexibility alone has already led to better results; introducing full flexibility of both the receptor and the ligand is a computationally difficult problem that is crucial to correctly modeling the molecular recognition process. A large number of academic groups are now actively involved in this field.

articles
Complexes of alkylene-linked tacrine dimers with Torpedo californica acetylcholinesterase: Binding of bis(5)-tacrine produces a dramatic rearrangement in the active-site gorge Rydberg, E.H., Brumshtein, B., Greenblatt, H.M., Wong, D.M., Shaya, D., Williams, L.D., Carlier, P.R., Pang, Y.-P., Silman, I. and Sussman, J.L. J. Med. Chem. 49 2006
eMovie: A storyboard-based tool for making molecular movies Hodis, E., Schreiber, G., Rother, K. and Sussman, J.L. Trends in Biochemical Sciences 32 2007
Developing Better Models of Flexibility¶
This chapter has presented a number of methods currently in development to increase the sampling rate for representing a greater portion of the conformational space of the receptor: combinatorial, stochastic, multiple protein structures are examples of methods currently under development. An entirely new generation of methods is however needed, that introduce a larger range of protein flexibility corresponding to simulations that are closer to reality.

articles
Protein-induced Conformational Changes of RNA During the Assembly of Human Signal Recognition Particle Elena Menichelli, Catherine Isel, Chris Oubridge and Kiyoshi Nagai J. Mol. Biol. 367 2007 10.1016/j.jmb.2006.12.056
Importance of Visual Docking¶
Although we focused in this chapter on computer-based calculations, it is important to note that molecular visualizations have played an important role in the development of molecular docking algorithms. Manual docking continues to be of practical use in the pharmaceutical industry and can still spark intuition and guide scientists towards new ideas or other strategies.

Media
This picture was is a snapshot of the VMD software VMD
Requirement for Manual Docking¶
A mandatory requirement of manual docking is an initial binding hypothesis. For example if the binding mode of a closely related ligand is already known even on a related target, this can be exploited. In the protein kinase field the bidentate recognition of ATP by the kinases can be exploited in kinase inhibition.

Illustration of Manual Docking¶
In a study aiming at finding inhibitors of the Akt1 protein kinase, Forino et al. found that by simply relying on results provided by different docking programs such as FlexX, Gold or CSCORE, the hit rate was slightly superior to the one expected from a random approach (~0.01-0.5%). However, by combining the docking programs with structural elements (H-bonds) that were found experimentally in a complex with Akt1, the authors were able to achieve a hit rate of 10%.

articles
Virtual docking approaches to protein kinase B inhibition Forino M. et Al. J. Med. Chem. 48 2005
Manual Docking with Solid Models¶
Manual docking can be also done using solid models. Such objects give researchers a feel of how the molecules fit together, or enable them to identify an initial crude pose to be further refined with computer graphics. However this approach has little practical value. The rigid objects shown below correspond to models of Fasciculin (right) and AChE (left).

Media
This picture was produced by the Helisys laminated object manufacturing equipment from the 3D coordinates of the two molecules.
Virtual Reality Docking System¶
Taking advantage of recent developments in virtual reality technology, virtual environment systems have emerged allowing the researcher to interactively perform molecular docking experiments. CAVE is an example of such a system developed in 1991 at the University of Illinois. Adapted to docking purposes it provides the illusion of three-dimensional immersion by projecting stereo images on the walls and floor of a room-sized cube. As the user moves, the new perspective is displayed in real-time to generate a fully immersive experience.

articles
An interactive system for virtual molecular docking Levine, D. et al. IEEE Comput. Sci. Eng. 4(2) 1997 10.1109/99.609834
applying virtual reality visualization to protein docking and design Anderson A. Journal of Molecular Graphics and Modelling 17(3) 1999 10.1016/S1093-3263(99)00029-7
book
Cruz-Neira C. et al. In Proceedings of the ACM SIGGRAPH Computer Graphics Conference
1993
Example of Docking using CAVE¶
The image shown below is the cAMP-dependent protein kinase (green) binding with an inhibitor molecule (red). The binding site is shown as a mesh of atoms and bonds (red), and the inhibitor itself is hovering above the binding site, ready to be docked. The docking is controlled using the CAVE wand (yellow), operated by a joystick device controlled manually by the user.

Synergy Between Interactive & Automated Docking¶
The combination of virtual reality (VR) interactive docking and automated docking algorithms gives researchers unique tools. VR docking stimulates creativity and intuition by giving a concrete sense of the 3D context. On the other hand, automated docking guides the user in his interactive virtual exploration, or lets him take control ("automated piloting") to propose a new solution or refine an existing one.

articles
applying virtual reality visualization to protein docking and design Anderson A. Journal of Molecular Graphics and Modelling 17(3) 1999 10.1016/S1093-3263(99)00029-7
Interactive Computer-Guided Docking¶
One example of an interactive computer-guided docking system involves force-feedback capabilities where the strength of the forces exerted are physically felt by the user. For example these forces can correspond to the Van der Waals force-field component of the energy of the complex. This enables the user to intuitively react and progressively guide docking in the active site.

articles
Interactive Computer-Aided Design for Molecular Docking and Assembly Lai-Yuen S.K., and Lee Y.S. Computer-Aided Design and Applications 3(6) 2006
Concept and prototype of protein-ligand docking simulator with force feedback technology Nagata H, et al. Bioinformatics 18 2002
Protein-Protein Docking Benchmarks¶
An important facet in the development of docking programs is the ability to measure the validity of a given algorithm, and test it in diverse situations. For this purpose a benchmark of 84 protein-protein complexes has been drawn up for which at least one of the unbound components is available in 3D, for testing docking methods and evaluating their progress. They contain various types of interactions as indicated in the diagram below and are classified into three groups: rigid body, medium and difficult complexes.

articles
A Protein-Protein Docking Benchmark, Proteins Proteins Chen R, Mintseris J, Janin J, Weng Z Proteins: Structure, Function, and Genetics 52 2003
Protein-Protein Docking Benchmark 2.0:an Update. Mintseris, J., Wiehe, K., Pierce, B., Anderson, R., Chen, R., Janin, J. and Weng, Z. Proteins: Structure Function and Genetics 60 2005
The CAPRI Competition¶
A key component that has boosted the development of reliable protein-protein docking methods is the CAPRI challenge involving developers of new methods. CAPRI (Critical Assessment of PRedicted Interactions) started in 2001 and is held approximately twice a year. Its goal is to assess the capacity of theoreticians to correctly predict protein-protein complexes.

articles
CAPRI: A Critical Assessment of PRedicted Interactions Janin J. et al. Proteins: Structure Function and Genetics 52 2003 10.1002/prot.10381
Welcome to CAPRI: A Critical Assessment of PRedicted Interactions Joel Janin Proteins: Structure, Function, and Bioinformatics 47(3) 2002 10.1002/prot.10111
wikipedia
Six Weeks for Submitting Predicted Complexes¶
A CAPRI round starts when a complex not present in the PDB is solved by a crystallographer and is donated to the CAPRI experiment. This complex is suitable for the CAPRI experiment if at least one structure of the interacting partners is available in the unbound form. The participants are given the structures of the interacting proteins and 10 candidate complexes are submitted by each group. In some cases, the homologs can be given, so that the participants are required to use homology modeling.

Assessment of the Predictions¶
The submissions are evaluated by comparison with the X-ray coordinates of the complexes, communicated prior to publication by the crystallographers. A successful docking requires the generation of a set of predictions which includes at least one near the correct solution. Each participant is allowed to submit five predicted models of each target complex.
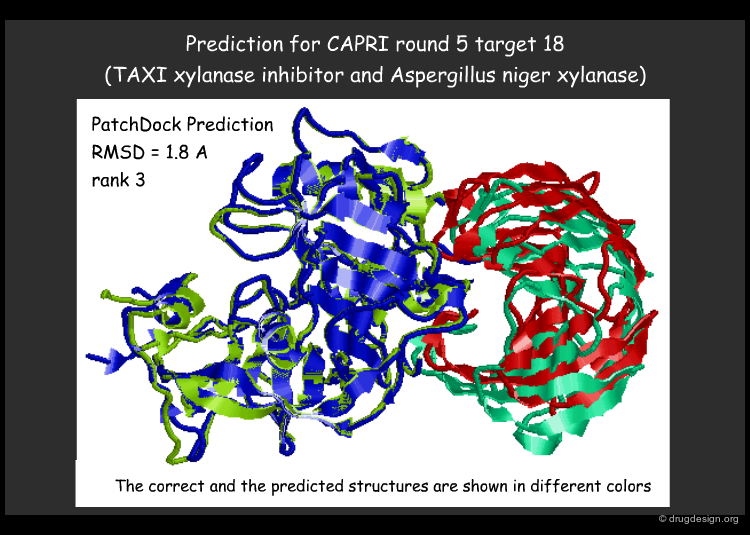
A New CAPRI Scoring Category¶
A scoring category was recently added to CAPRI to assess the ability of scoring function developers to identify correct protein-protein complexes. Sets of 100 candidate poses are generated by theoreticians and submitted to scorers. Each scorer studies the theoretical complexes and picks the 10 structures for which he obtained the highest scores. The submissions are evaluated by comparison with the experimental data provided by the crystallographers.

CAPRI History and Experience¶
CAPRI round one began in 2001 with three target protein-protein complexes; two months later, 271 predictions had been submitted. The CAPRI rounds take place approximately every 6 months with about 1-6 target protein-protein complexes. 19 groups of predictors participated in the first round and 40 in round eleven (2006). As stressed by the Baker group, an important lesson learned from the CAPRI experiments is that the current bottleneck for developing protein docking methods is how to treat backbone flexibility, as there were almost complete failures for the targets with backbone movements upon forming the complex.

Perspectives¶
Despite the current limitations described here, computational docking has made tremendous progress in the last few years and is continuously growing as a new and recognized discipline. Computational docking is currently routinely used in drug discovery projects in the pharmaceutical industry. Success stories are numerous, some of which are listed below.



articles
Thienothiopyran-2-sulfonamides: novel topically active carbonic anhydrase inhibitors for the treatment of glaucoma John J. Baldwin, Gerald S. Ponticello, Paul S. Anderson, Marcia E. Christy, Mark A. Murcko, William C. Randall, Harvey Schwam, Michael F. Sugrue, Pierre Gautheron, et al. J. Med. Chem. 32(12) 1989
Rational Design of Potent Sialidase-based Inhibitors of Influenza Virus Replication von Itzstein, M.; Wu, W. Y.; Kok, G. B.; Pegg, M. S.; Dyason, J. C.; Jin et al. Nature 363 1993
Amprenavir: a new human immunodeficiency virus type 1 protease inhibitor Fung,H.B., Kirschenbaum,H.L. and Hameed,R. Clin Ther 22 2000
Structure-based design of Aliskiren, a novel orally effective renin inhibitor Jeanette M. Wood, Juergen Maibaum, Joseph Rahuel, Marcus G. Gruetter, Nissim-Claude Cohen, Vittorio Rasetti, Heinrich Rueger, Richard G?schke, Stefan Stutz, Walter Fuhrer, Walter Schilling, Pascal Rigollier, Yasuchika Yamaguchi, Frederic Cumin, Hans-Peter Baum, Christian R. Schnell, Peter Herold, Robert Mah, Chris Jensen, Eoin O'Brien, Alice Stanton and Martin P. Bedigian Biochemical and Biophysical Research Communications 308 2003
Crystal Structure of Escherichia coli Thymidylate Synthase Containing Bound 5-Fluoro-2'-Deoxyuridylate and 10-Propargyl-5,8-Dideazafolate Matthews DA, Appelt K, Oatley SJ and Xuong NH J. Mol. Biol. 214 1990
Structure, Multiple Site Binding, and Segmental Accommodation in Thymidylate Synthase on Binding 5- FdUMP and an Anti-folate Montfort WR, Perry KM, Fauman EB, Finer-Moore JS, Maley GF, Hardy L, Maley F and Stroud RM Biochemistry 29 1990
Viracept (Nelfinavir mesylate, AG 1343): a potent orally bioavailable inhibitor of HIV-1 protease Kaldor SW, Kalish VJ, Davies JF, Shetty BV, Fritz JE, Appelt, K, Burgess JA, Campanale KM et al. J Med Chem 40 1997
Potent and Selective Inhibitors of the ABL-Kinase: Phenylamino-pyrimidine (PAP) Derivatives J. Zimmermann et al. Bioorganic and Medicinal Chemistry Letters 7 (2) 1997
Antiviral properties of Ro 31-8959, an inhibitor of human immunodeficiency virus (HIV) proteinase Craig JC, Duncan IB, Hockley D, Grief C, Roberts NA, Mills JS Antiviral Res 16(4) 1991
L-735,524: An Orally Bioavailable Human Immunodeficiency Virus Type 1 Protease Inhibitor JP Vacca, BD Dorsey, WA Schleif, RB Levin, SL McDaniel, PL Darke, J Zugay, JC Quintero, OM Blahy, E Roth, VV Sardana, AJ Schlabach, PI Graham, JH Condra, L Gotlib, MK Holloway, J Lin, I Chen, K Vastag, D Ostovic, PS Anderson, EA Emini, and JR Huff Proc. Natl. Acad Sci. USA 91 1994
Discovery of Ritonavir, A potent inhibitor of HIV protease with high oral bioavailability Kempf DJ, Sham HL, Marsh KC, Flentge CA, Betebenner D, Green BE, McDonald E, Vasavanonda S, Saldivar A, Wideburg NE, Kati WM, Ruiz L, Zhao C, Fino LM, Patterson, J, Molla A, Plattner JJ, Norbeck DW and clinical efficacy. J Med Chem 41 1998
Oxyguanidines - part 2: Discovery of a novel orally active thrombin inhibitor through structure-based drug design and parallel synthesis Lu, T., Markotan, T., Coppo, F., Tomczuk, B., Crysler, C., Eisennagel, S., Spurlino, J., Gremminger, L., Soll, R.M., Giardino, E.C., Bone, R. Oxyguanidines BIOORG.MED.CHEM.LETT. 14 2004
Inhibitors of Multiple Mutants of Plasmodium falciparum Dihydrofolate Reductase and Their Antimalarial Activities Sumalee Kamchonwongpaisan, Rachel Quarrell, Netnapa Charoensetakul, Rachel Ponsinet, Tirayut Vilaivan, Jarunee Vanichtanankul, Bongkoch Tarnchompoo, Worachart Sirawaraporn, Gordon Lowe, and Yongyuth Yuthavong J. Med. Chem. 47 2004
A new class of inhibitors of human leucocyte elastase Hassall CH, Johnson WH, Kennedy AJ, Roberts NA. FEBS Lett 183(2) 1985
Inhibitors of the enzyme purine nucleoside phosphorylase Morris PE, Montgomery JA Exp Opin Ther Patents 8 1998
Structure-based design of potent, amidine-derived inhibitors of factor Xa: evaluation of selectivity, anticoagulant activity, and antithrombotic activity Wiley MR, Weir LC, Briggs S, Bryan NA, Buben J, Campbell C, Chirgadze NY, Conrad RC, Craft TJ, Ficorilli JV, Franciskovich JB, Froelich LL, Gifford-Moore DS, Goodson T Jr, Herron DK, Klimkowski VJ, Kurz KD, Kyle JA, Masters JJ, Ratz AM, Milot G, Shuman RT, Smith T, Smith GF, Tebbe AL, Tinsley JM J Med Chem 43(5) 2000
Discovery of a Potent Small Molecule IL-2 Inhibitor through Fragment Assembly Andrew C. Braisted, Johan D. Oslob, Warren L. Delano, Jennifer Hyde, Robert S. McDowell, Nathan Waal, Chul Yu, Michelle R. Arkin, and Brian C. Raimundo J. AM. CHEM. SOC. 125 2003
The design and synthesis of sulfonamides as caspase-1 inhibitors William G. Harter, Hans Albrect, Kenneth Brady, Bradley Caprathe, James Dunbar, John Gilmore, Sheryl Hays, Catherine R. Kostlan, Beth Lunneya and Nigel Walkerb Bioorganic and Medicinal Chemistry Letters 14 2004
Imidazo[1,2-a]pyridines: A Potent and Selective Class of Cyclin-Dependent Kinase Inhibitors Identified Through Structure-Based Hybridisation Malcolm Anderson, John F. Beattie, Gloria A. Breault, Jason Breed, Kate F. Byth, Janet D. Culshaw, Rebecca P. A. Ellston, Stephen Green, Claire A. Minshull, Richard A. Norman, Richard A. Pauptit, Judith Stanway, Andrew P. Thomas and Philip J. Jewsbury Bioorganic and Medicinal Chemistry Letters 13 2003
Structural Determinants of CDK4 Inhibition and Design of Selective ATP Competitive Inhibitors Campbell McInnes, Shudong Wang,Sian Anderson, Janice O'Boyle, Wayne Jackson, George Kontopidis, Christopher Meades, Mokdad Mezna, Mark Thomas, Gavin Wood, David P. Lane, and Peter M. Fischer Chemistry and Biology 11 2004
CH...O and CH...N Hydrogen Bonds in Ligand Design: A Novel Quinazolin-4-ylthiazol-2-ylamine Protein Kinase Inhibitor Albert C. Pierce, Ernst ter Haar, Hayley M. Binch, David P. Kay, Sanjay R. Patel, and Pan Li J. Med. Chem. 48 2005
Discovery of a Potent, Selective Protein Tyrosine Phosphatase 1B Inhibitor Using a Linked-Fragment Strategy Bruce G. Szczepankiewicz, Gang Liu, Philip J. Hajduk, Cele Abad-Zapatero,Zhonghua Pei, Zhili Xin, Thomas H. Lubben, James M. Trevillyan, Michael A. Stashko, Stephen J. Ballaron, Heng Liang, Flora Huang, Charles W. Hutchins, Stephen W. Fesik, and Michael R. Jirousek J. AM. CHEM. SOC. 125 2003
Novel Selective Small Molecule Agonists for Peroxisome Proliferator-Activated Receptor (PPAR-delta)-Synthesis and Biological Activity Marcos L. Sznaidman, Curt D. Haffner, Patrick R. Maloney, Adam Fivush, Esther Chao, Donna Goreham, Michael L. Sierra, Christelle LeGrumelec,b H. Eric Xu, Valerie G. Montana, Millard H. Lambert, Timothy M. Willson, William R. Oliver, Jr. and Daniel D. Sternbach Bioorganic and Medicinal Chemistry Letters 13 2003
Structure-Based Design of Potent Retinoid X Receptor alpha Agonists Curt D. Haffner, James M. Lenhard, Aaron B. Miller, Darryl L. McDougald, Kate Dwornik et al. J. Med. Chem. 47 2004
Structure-Based Design of Estrogen Receptor-beta Selective Ligands Eric S. Manas, Rayomand J. Unwalla, Zhang B. Xu, Michael S. Malamas, Chris P. Miller, Heather A. Harris, Chulai Hsiao, Tatos Akopian et al. J. AM. CHEM. SOC. 126 2004
A Highly Potent Non-Nucleoside Adenosine Deaminase Inhibitor: Efficient Drug Discovery by Intentional Lead Hybridization adashi Terasaka, Takayoshi Kinoshita, Masako Kuno, and Isao Nakanishi J. AM. CHEM. SOC. 126 2004
Structure-Based Design and Synthesis of Non-Nucleoside, Potent, and Orally Bioavailable Adenosine Deaminase Inhibitors Tadashi Terasaka, Hiroyuki Okumura, Kiyoshi Tsuji, Takeshi Kato, Isao Nakanishi, Takayoshi Kinoshita, Yasuko Kato, Masako Kuno, Nobuo Seki, Yoshinori Naoe, Takeshi Inoue, Kohichiro Tanaka, and Katsuya Nakamura J. Med. Chem. 47 2004
Influenza Neuraminidase Inhibitors: Structure-Based Design of a Novel Inhibitor Series Vincent Stoll, Kent D. Stewart, Clarence J. Maring, Steven Muchmore et al. Biochemistry 42 2003
Design, synthesis, and activity of 4-quinolone and pyridone compounds as nonthiol-containing farnesyltransferase inhibitors Qun Li, Akiyo Claiborne, Tongmei Li, Lisa Hasvold, Vincent S. Stoll et al. Bioorganic and Medicinal Chemistry Letters 14 2004
Design of HIV-1 Protease Inhibitors Active on Multidrug-Resistant Virus Dominique L. N. G. Surleraux, Herman A. de Kock, Wim G. Verschueren et al J. Med. Chem. 48 2005
Structure-based design of potent and selective inhibitors of collagenase-3 (MMP-13) Soong-Hoon Kim, Andrew T. Pudzianowski, Kenneth J. Leavitt et al. Bioorganic and Medicinal Chemistry Letters 15 2005
Entry into a New Class of Potent Proteasome Inhibitors Having High Antiproliferative Activity by Structure-Based Design Pascal Furet, Patricia Imbach, Maria Noorani, Juergen Koeppler, Kurt Laumen, Marc Lang, Vito Guagnano, Peter Fuerst, Johannes Roesel, Johann Zimmermann, and Carlos Garcia-Echeverria J. Med. Chem. 47 2004
Structure-Based Design of Potent and Selective Cell-Permeable Inhibitors of Human beta-Secretase (BACE-1) Shawn J. Stachel, Craig A. Coburn, Thomas G. Steele, Kristen G. Jones, Elizabeth F. Loutzenhiser, Alison R. Gregro et al. J. Med. Chem. 47 2004
Potent and selective P2-P3 ketoamide inhibitors of cathepsin K with good pharmacokinetic properties via favorable P1', P1, and/or P3 substitutions David G. Barrett, John G. Catalano, David N. Deaton, Anne M. Hassell et al. Bioorganic and Medicinal Chemistry Letters 14 2004
Design, Parallel Synthesis, and Crystal Structures of Pyrazinone Antithrombotics as Selective Inhibitors of the Tissue Factor VIIa Complex John J. Parlow, Brenda L. Case, Thomas A. Dice, Ricky L. Fenton, Michael J. Hayes et al. J. Med. Chem. 46 2003
A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design GL Card, L Blasdel, BP England, C. Zhang et al. Nature Biotechnology 23(2) 2005
Structure-based design of substituted diphenyl sulfones and sulfoxides as lipophilic inhibitors of thymidylate synthase Jones TR, Webber SE, Varney MD, Reddy MR, Lewis KK,Kathardekar V, Mazdiyashni H, Deal J, Nguyen D, Welsh KM, Webber S, Johnston A, Matthews DA, Smith WW, Janson, CA, Bacquet RJ, Howland EF, Booth CLJ, Herrmann SM, Ward RW, White J, Bartlett CA, Morse CA J Med Chem 40 1997
Copyright © 2024 drugdesign.org