Molecular Similarity¶
Info
"Molecular similarity" is a widely applied concept in computer-aided drug design, and is used for both the identification of novel active compounds or compounds with specific properties (an approach often called "virtual screening") as well as the replacement of unfavorable functional groups ("bioisosteric replacement"). While the underlying principle that "similar molecules tend to have similar properties" has been around since the early days of medicinal chemistry, it has recently received renewed interest with the large-scale application of computers in drug design and discovery. What defines similarity in the world of molecules is discussed in this chapter.
Number of Pages: 140 (±3 hours read)
Last Modified: January 2006
Prerequisites: None
Introduction¶
Similarity and Complementarity-Based Drug Design¶
Ligand-based and structure-based drug design are the two major approaches in rational drug design. Ligand-based drug design exploits the likeness between molecules that is expressed in terms of "molecular similarity" whereas structure-based drug design exploits the detailed 3D recognition features between a ligand and its receptor, and the concept used is "molecular complementarity". The molecular similarity concept and its applications are discussed in detail in this chapter.

Comparing Molecules: a Central Issue in Drug Discovery¶
Assessing the similarity between molecules is a central issue in drug discovery. The molecular similarity concept has created a broad range of cheminformatics tools that have proven useful in drug design for finding new lead compounds.
The Molecular Similarity Principle¶
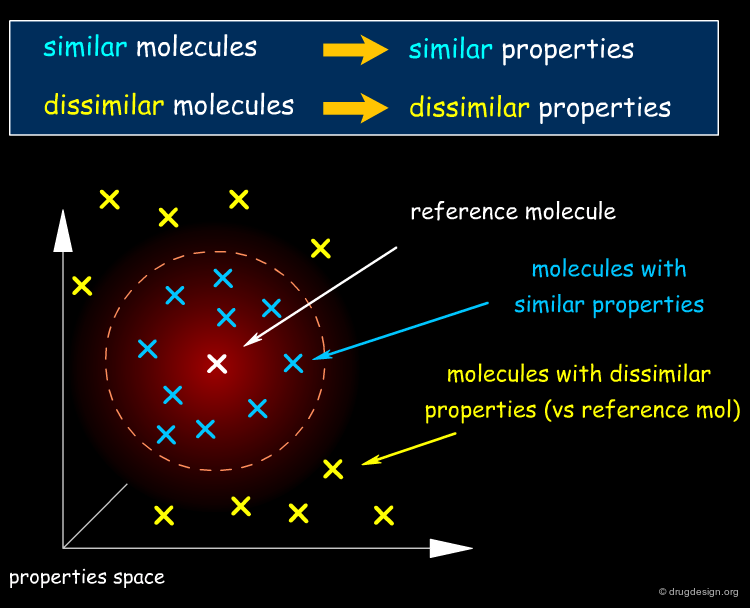
The "molecular similarity principle", which is also known as the "similar property principle", is the underlying concept of virtually all ligand-based drug design methods. Its underlying assumption states that similar molecules tend to behave similarly, while more dissimilar molecules exhibit more distinct properties.
articles
Chemical Similarity Searching P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
Approaches to Measure Chemical Similarity N. Nikolova and J. Jaworska. QSAR Comb. Sci. 22 2004 10.1002/qsar.200330831
Molecular similarity: a key technique in molecular informatics A. Bender and R.C. Glen Org. Biomol. Chem. 2 2004 10.1039/b409813g
book
M.A. Johnson and G.M. Maggiora. Concepts and Applications of Molecular Similarity Wiley 1990
Subjectivity of the Similarity Concept¶
Similarity is a subjective concept that always requires a definition of the context in which it is used. For example, an eagle and a sparrow are similar in the sense that they are both birds and thus able to fly. With respect to their sizes and weights however, they exhibit major differences. This idea is illustrated in the two similar patterns shown below: when examined in a horizontal orientation, judgments of differences are slow and tedious; this changes completely when traces are viewed with 90o clockwise rotation.

What can be Similar in Molecules?¶
Similarity depends on perspective: the same thing can be seen differently, depending on the context. Examples of differing molecular similarity perceptions induced by different 'filters' are shown in the view below and are discussed in the following pages.

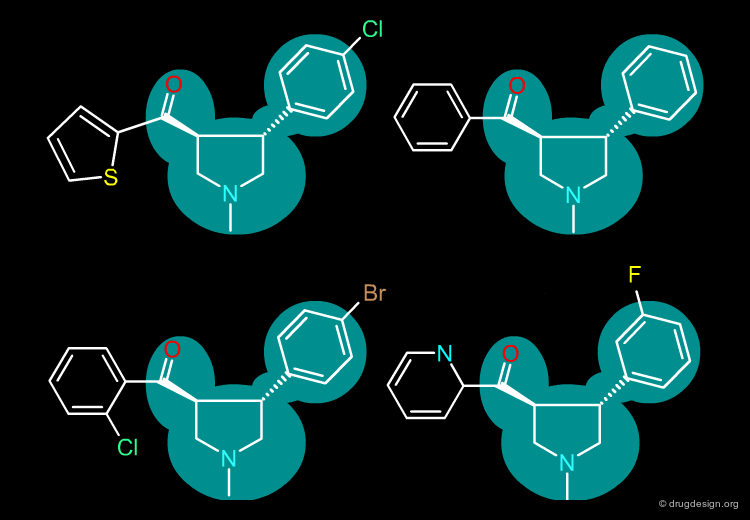
2D-Structure Similarity¶
Since chemists are very familiar with structural chemical formulas, a straightforward way to compare structures is based on their 2D connectivity i.e. the kinds of atoms which are bonded to each other, in which topology. In the structure below there are four structures which present identical bioactivity, and they possess very similar 2D structures.
Shape Similarity¶
Another way to define similarity between molecules is shape similarity. Molecular shape is an important determinant in the biological activity of a molecule. Below two structures are shown which look very different with respect to their 2D similarity. Still, if they are rendered in three dimensions you can see that their shapes are similar, which accounts for their similar biological profile.


Surface Physicochemical Similarity¶
Apart from shape, surface property similarity is very important. Properties such as atomic charges, electrostatic potentials, hydrophobicity, polarizability can be represented and compared on the molecular surfaces. Below you can see the surface electrostatic potential of two molecules: a catechol and its structurally quite dissimilar bioisosteric replacement containing a second nitrogen heterocycle instead of the two original hydroxyl groups. Despite the dissimilarity with respect to their 2D representations, both structures show similar electrostatic potential on the surface, which in turn might result in similar bioactivity.


articles
Isosterism and molecular modification in drug design C.W. Thornber Chem. Soc. Rev. 8 1979
book
The Organic Chemistry of Drug Design and Drug Action Academic Press 2004
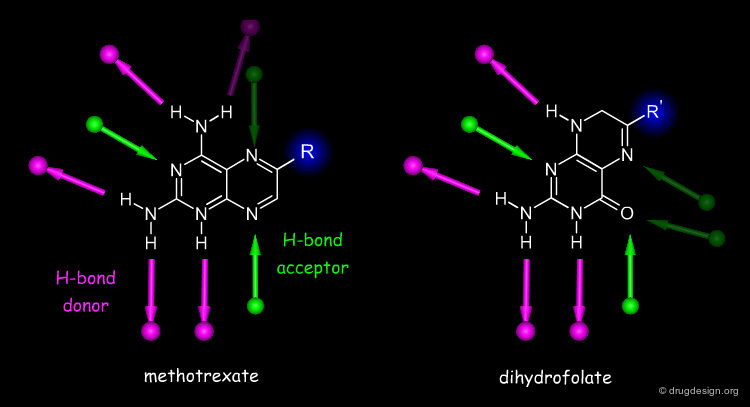
H-Bond Similarity¶
Since the hydrogen bonds are generally essential for the selectivity of a ligand, molecules can be compared in terms of their hydrogen bond pattern similarity. Both methotrexate and dihydrofolate bind to dihydrofolate reductase; by looking at their chemical structures it is reasonable to assume that they bind in an orientation as indicated in '2D alignment'. However, X-ray studies reveal that each molecule binds upside down, relative to the other, which makes sense when looking at their H-bond similarities (see 'H-bond alignment').


articles
What can we learn from molecular recognition in protein-ligand complexes for the design of new drugs? Bohm, H.J., Klebe, G. Angew. Chem. Int. Ed. Engl. 35 1996
Absence of Particular Features¶
Are a turtle and an elephant similar because neither of them do not have wings? While this is partly a philosophical question it needs to be mentioned since there are molecular similarity methods that, in addition to judging similarity based on features that indeed do match, also take features into account which do not match. The molecules shown below are all antidepressants. NK-1145 and LU-3010 do not have the tricyclic moiety normally found in the other structures, so in this context they are similar.
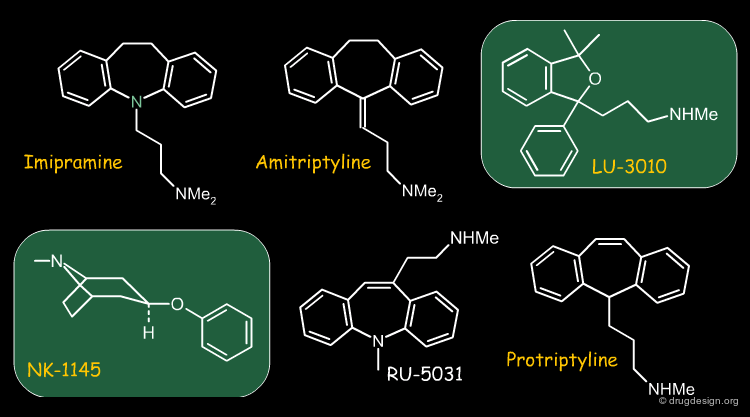
articles
Chemical Similarity Searching. P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
Pharmacophore Similarity¶
The pharmacophore is the three-dimensional (spatial) arrangement of ligand features responsible for ligand-target interactions. Comparing molecules in terms of their pharmacophore pattern focuses on considering only the essential parts of these molecules. In the example below the pharmacophore consists of a fluoro-phenyl, an amino group and a carboxyl moiety.


Comparing Molecular Characteristics¶
As illustrated, molecules can be compared in a multitude of ways, by taking into account for example connectivity properties, surface properties, shape of structures or putative interaction types in space. The characteristics that can be used to compare molecules are virtually infinite. But which characteristics should then be used to compare molecules in molecular similarity calculations?

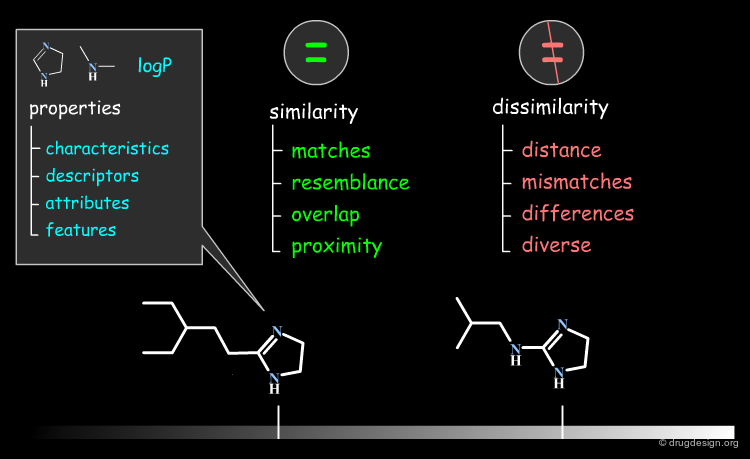
Terminology: Similarity Attributes¶
The vocabulary used for describing similarities or differences between objects can vary from one group to another. Current terminologies for properties are: characteristics, descriptors, attributes, features; for similarities: matches, resemblance, overlap, proximity; and for dissimilarities: distance, mismatches, differences and diverse.
Relevant Characteristics: What is Important?¶
The ideal characteristic used for a comparison should be like an ideal witness in court, who states "the truth, the whole truth, and nothing but the truth". In other words, the characteristic captures all the relevant aspects of the property we are attempting to predict ("the whole truth"), but at the same time does not add noise ("nothing but the truth"). Specific descriptors can be good or bad, depending on the situation.


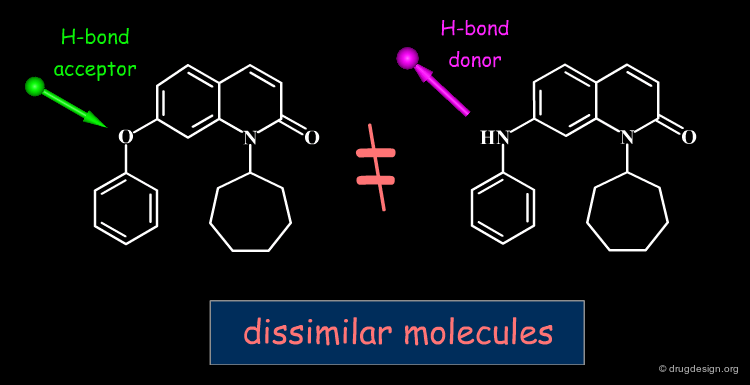
Relativity of Relevant Properties¶
In molecular similarity calculations the relevant descriptors differ from case to case. In the example below: in terms of lipophilicity the replacement of an oxygen linker (-O-) by a secondary amine (-NH-) does not introduce major changes; however this modification may have radical repercussions if the group is involved in specific hydrogen bond interactions with the receptor.

Interpretable Characteristics¶
It is a good rule of thumb to aim for properties that are interpretable because they provide insights into the exact content of the molecules at hand. However they only capture what chemists are trained to capture. Hard-to-interpret descriptors try to capture alternative aspects of the molecular structure that are elusive to the human mind, but can be related to observable properties.

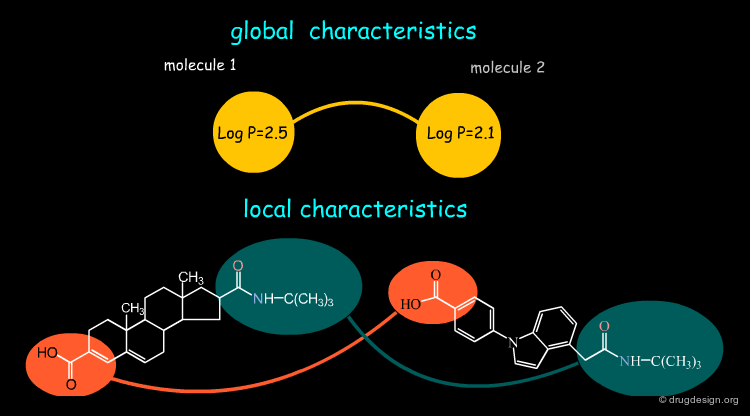
Global and Local Characteristics¶
Some molecular characteristics are 'global' and provide a very short description of the molecule, as for example the LogP. Other characteristics can be 'local', describing the properties of some regions, fragments, atoms or even a point in space. Local regions can be compared individually, enabling local similarities.
Maximizing Similarity: Object Alignments¶
The easiest way to maximize and reveal similarities between objects is to align them in a canonical way, where the objects are defined within the same referential. Molecular alignments are widely used for 1D, 2D, 3D comparisons, and beyond. In bioinformatics for example, sequences of genes or proteins are aligned to identify similarities and differences. In the example below 3D superimpositions are used to compare the two molecules (buttons '3D Before' and '3D After').


The Psychology of Similarity¶
Similarity is not a concept that is only present in computational drug design. Rather it has enormous influence on our everyday lives, for example identifying friends and relatives: while they may be wearing new clothes or have taken off their glasses, we are still able to recognize them. While this seems trivial, it is not that trivial to computers where every assumption has to be made explicit. For example, in the figure below you are easily able to identify a tree and distinguish it from other objects. But how would you write a computer algorithm to do that?

articles
Features of similarity A.Tversky Psychol. Rev. 84 1977
Molecular Similarity in Medicinal Chemistry Era¶
The concept of "molecular similarity" has been implicitly employed for many years by medicinal chemists. This will be presented in the next section with examples such as chemical modification, bioisosteric replacement, molecular mimicry, me-too drugs, peptidomimetics, lead-like and drug-like molecules.
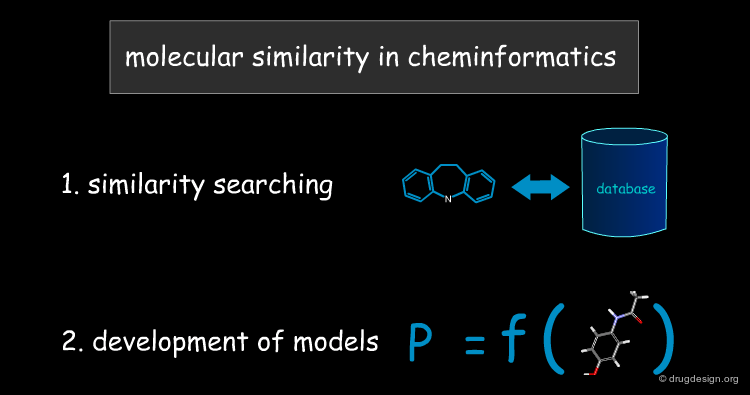
Cheminformatics¶
With the advent of computers, the definition of "molecular similarity" became more explicit, and two major approaches have emerged. The first is involved in similarity searching as the database implementation of the similarity concept. The second deals with the development of models for predicting molecular properties. Each of these approaches will be discussed in detail in the present chapter.
Medicinal Chemistry Approaches Based on the Similarity Principle¶
Chemical Modifications¶
The systematic chemical modification of a lead structure by making analogs without changing the chemical scaffold of the reference compound is a widely used approach in medicinal chemistry. It is guided by the molecular similarity principle and aims at finding a better compound with improved properties.

Bioisosteric Replacements¶
Bioisosteric replacement means the replacement of undesired functional groups with functionally similar, but structurally different "bioisosteres". Functional groups may be undesired for a multitude of reasons, including metabolic instability. In the example below two acidic groups were replaced by equivalent bioisosteres.

articles
Bioisosteres of 9-carboxymethyl-4-oxo-imidazo[1,2-a]indeno-[1,2-e]pyrazin-2-carboxylic acid derivatives. Progress towards selective, potent in vivo AMPA antagonists with longer durations of action. Jimonet P, et al. Bioorg Med Chem Lett. 11(2) 2001 10.1016/S0960-894X(00)00592-8
Molecular Mimicry¶
The analysis of the similarities and dissimilarities between active and inactive molecules gives a "feeling" of how structural variations can change biological properties and helps recognize the structural elements that are important for biological activities. Molecular mimicry consists of introducing these elements in an entirely new molecule, which is chemically unrelated to the structure of the known reference compounds (at least in part).
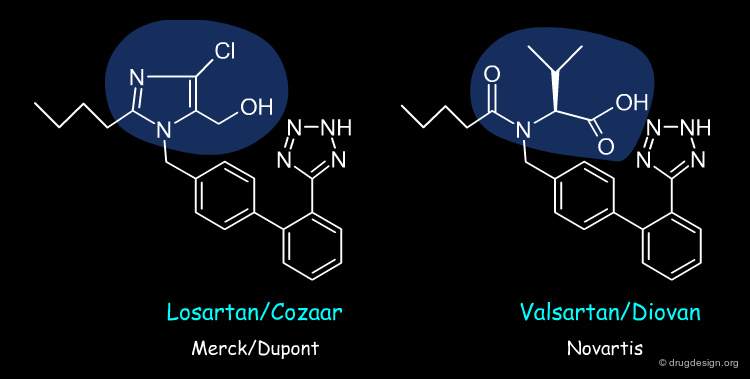
articles
Nonpeptide angiotensin II receptor antagonists: the discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives David J. Carini, John V. Duncia, Paul E. Aldrich, Andrew T. Chiu, Alexander L. Johnson, Michael E. Pierce, William A. Price, Joseph B. Santella, III, Gregory J. Wells, and et al. J.Med.Chem.
1991
Nonpeptide Angiotensin II Receptor Antagonists: The Next Generation in Antihypertensive Therapy Ruth R. Wexler, William J. Greenlee, John D. Irvin, Michael R. Goldberg, Kristine Prendergast, Ronald D. Smith, and Pieter B. M. W. M. Timmermans J.Med.Chem. 39 1996
Valsartan, a potent, orally active angiotensin II antagonist developed from the structurally new amino acid series Peter Buehlmayer, Pascal Furet, Leoluca Criscione, Marc de Gasparo, Steven Whitebread, Tibur Schmidlin, Rene Lattmann and Jeanette Wood Bioorg. Med. Chem. Lett 4 (1) 1994
Pharmacological profile of valsartan: a potent, orally active, nonpeptide antagonist of the angiotensin II AT1-receptor subtype. Criscione L, et al. Br J Pharmacol. 110(2) 1993
Mee-too-ism¶
It is common practice in the pharmaceutical industry to take an existing drug or a drug under development and introduce a small change in its chemistry. Such copycat drugs are usually called me-too drugs in industry jargon, and their development is considered to be motivated by purely commercial considerations. Notwithstanding such perceptions, historically, many "me-too" drugs have proved to be considerably better than their original counterparts. Here also molecular similarity is the driving principle aimed at keeping most of the structure of the original drug intact, while looking for a small structural difference that will provide an important new advantage.

articles
The discovery and preparation of disubstituted novel amino-aryl-piperidine-based renin inhibitors Cody WL, et al. Bioorg Med Chem. 13 2005
Peptidomimetics¶
Peptidomimetics are compounds that are peptide mimetics. They have non-peptide structure in part or in full, they are structural mimics of the reference peptides and retain their pharmacological activities. As presented and illustrated in the several chapters devoted to peptidomimicry in this courseware, the design of such compounds is guided by an intelligent exploitation of molecular similarity principles. Below is shown an example of peptidomimetics.
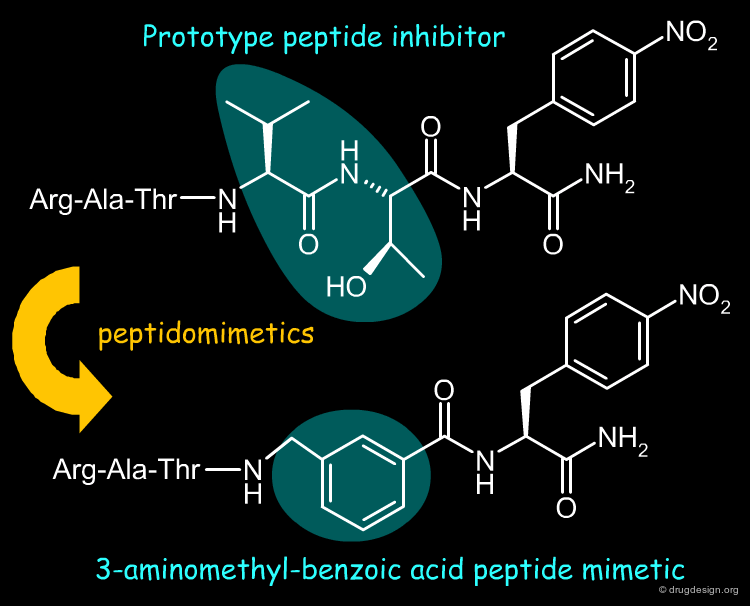
articles
Peptides to peptidomimetics: towards the design and synthesis of bioavailable inhibitors of oligosaccharyl transferase Weerapana E, Imperiali B. Org Biomol Chem. 1 2003 10.1039/b209342a
Lead-Like and Drug-Like Approaches¶
Chemists can now build and measure the 'lead-like' or 'drug-like' properties of molecules (concepts presented in another chapter). These provide useful structure-property relationships that complement current structure-activity information. These properties derive directly from molecular similarity structural analyses.
articles
A Scoring Scheme for Discriminating Between Drugs and Non-Drugs J. Sadowski and H. Kubinyi, J. Med. Chem. 41 1998
Classifying 'Drug-likeness' with Kernel-Based Learning Methods K.R. Muller et al. J. Chem. Inf. Model. 45 2005
Similarity Searching in Database¶
Exact and Substructure Searching¶
Until the mid 1980s database searching was limited to exact and substructure search, i.e. the identification of all the molecules in the database that contain a specified substructure. Substructure searching is based on sub-graph algorithms that are computationally lengthy.

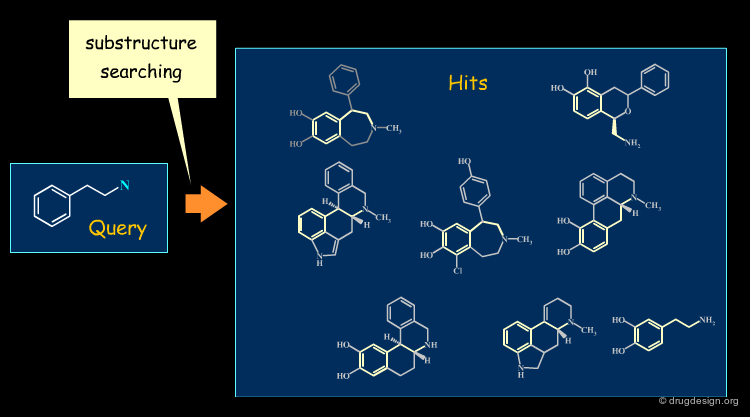
Similarity Searching¶
The implementation of similarity principles in database searching was first introduced in the late 1980s. While substructural searching attempts exact retrieval of a certain substructure, in similarity searching, compounds (or parts of compounds) with different substructures but similar physico-chemical properties are identified.

Semi-Manual Similarity Searching¶
Note that the medicinal chemist did not wait for advanced computerized programs to start searching for similar molecules to a reference compound with different sub-structural patterns. For example, starting from a reference compound, the chemist imagined several possible chemical modifications he explored and analyzed by traditional substructural searching. This is a cumbersome and sometimes discouraging process (low hit rate). Moreover, the approach is biased by the chemist's imagination and is far from being systematic.
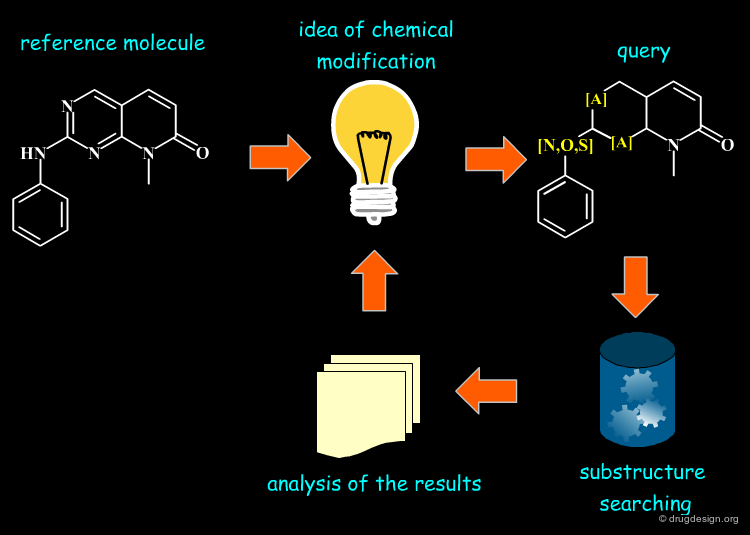
Similarity Concept and Creativity¶
Since similarity searching is a fuzzy concept, the medicinal chemist does not know exactly what he is looking for. This in fact enables the chemist to extend and control creativity by focusing on new ideas and concepts.
Output of Similarity Searching¶
One of the big advantages of similarity searching is that the results are well structured, with scoring indicating the degree of similarity with the reference molecule. This makes it possible to analyze the results by order of importance. This is different from traditional substructure searches whose outputs were chaotic, consisting of the accumulation of results obtained from different sessions.

Broad Range of Applications¶
Because of its simplicity of use and a computational speed that can routinely compare millions of compounds in minutes, database similarity searching is widely used in a great number of applications. Some of these applications are listed below and will be presented later in more detail.

Substructure & Similarity Searching Complementarities¶
The emergence of similarity searching programs has not superseded substructure-searching techniques by making them obsolete; on the contrary the two techniques are complementary. For example substructural searching can be used to eliminate toxic compounds containing well defined toxicophores, whereas similarity searching is the best choice to find a more soluble scaffold. Another aspect of this complementarity is illustrated here, where the speed of the initial computer intensive substructure search algorithm was substantially improved by using a pre-filtering procedure, based on similarity searching.

General Requirements of a Method¶
Three steps are required for the development of a similarity searching computerized method: first, structures and properties have to be represented in a computer-understandable format. Second, a similarity (or dissimilarity) measure between those representations is required to obtain a numerical similarity value. And finally, a defined algorithm needs to be implemented to perform the desired task.

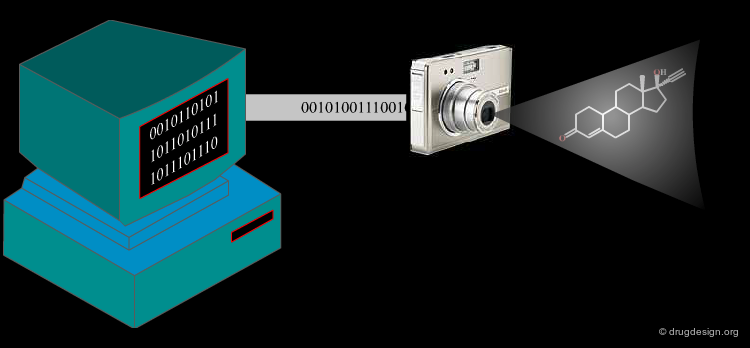
Make Molecules Accessible to the Computer¶
Computers are binary machines: the only digits they have are ones and zeros. In order to compare molecules, it is necessary to encode their structures and properties in a suitable representation. A molecular structure can be represented in the computer in its connectivity-table format, where atoms are represented as nodes of a graph and bonds as edges of it: this representation resembles what chemists draw as "chemical structures". Another representation is based on molecular descriptors. This representation is better suited to similarity searching because both the structures and the properties of the molecules can be described. Molecular descriptor methods will be presented in the next section.
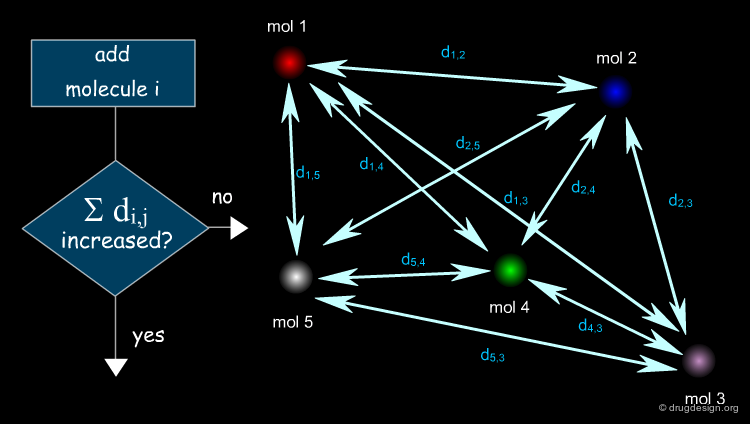
Need of Methods to Measure Similarity¶
If a computer-interpretable representation for a compound has been encoded, the "position" of the compound, or its coordinates , in "chemical space" are defined. In order to compare compounds, the "distance" between them has to be defined to obtain a numerical similarity value. Whatever the particular type of similarity measure (sometimes called 'distance'), its most important property is that the similarity it assigns to two structures must be related to the similarity of the properties of the molecules.

Apply an Algorithm¶
While the representation of structures as well as a measure of similarity is required by all molecular similarity computer-based methods, algorithms are needed to carry out the desired task. For example, one might wish to select a set of diverse compounds from a library. The algorithm will be directed to maximize the distances between the molecules selected.
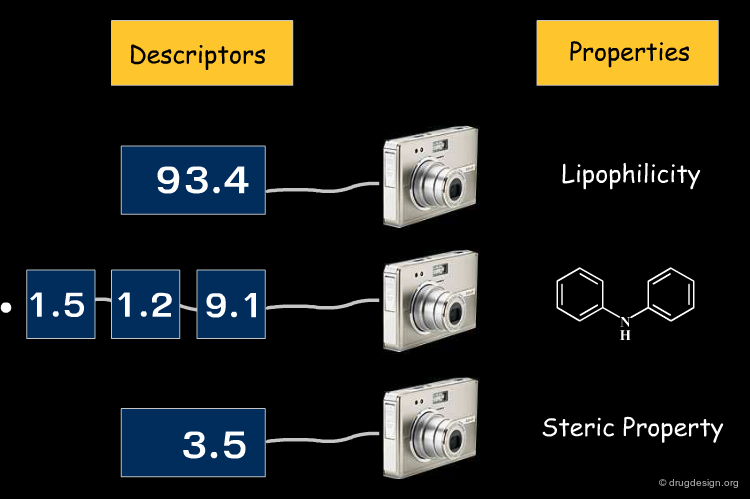
Molecular Descriptors: Make Molecules Accessible to the Computer¶
The Concept of "Molecular Descriptors"¶
Molecular descriptors consist of numbers that capture the properties and the structures of molecules. They then serve to characterize molecules and compare them by searching through large libraries with respect to physico-chemical, structural and biological properties. Molecular descriptors provide an abstract representation of molecular structures in "descriptor space". This means that structural molecular features are analyzed by an algorithm, and, while trying to capture information relevant to the problem, are translated into a structural format that can be further analyzed by the computer.
Selection of Relevant Descriptors¶
As already mentioned, it is crucial to identify properties that are relevant to the problem at hand. Molecular descriptors that capture these properties are called relevant descriptors. The "molecular similarity principle" is applicable only with relevant descriptors. In the figure below, A and B are bad descriptors, whereas X and Y are relevant ones.
![]()
High-Dimensionality Space of the Molecular Descriptors¶
In general many properties must be taken into account to improve the signal-to-noise ratio and several descriptors might be necessary to capture a single property. The "chemical space" will be characterized by a high degree of dimensionality.

articles
Feature selection in quantitative structure-activity relationships W.P. Walters and B.B. Goldman Curr. Opin. Drug Discov. Devel. 8 2005
Molecular Similarity Searching Using Atom Environments, Information-Based Feature Selection, and a Naive Bayesian Classifier A. Bender, et al., J. Chem. Inf. Comput. Sci. 44 2004 10.1021/ci034207y
Example of Selection of Relevant Descriptors¶
The example below deals with face recognition. The most straightforward way to recognize people is to compare individual pixels of the picture to each other. This procedure would however quickly run into problems if the face moved slightly while the picture was being taken or a change in lighting made the picture slightly lighter or darker. Thus, a computer selected characteristic feature from the faces below and the distances between them do not depend on the precise orientation of the face, and they are also insensitive to changes in illumination. They are relevant descriptors.

articles
Feature selection in quantitative structure-activity relationships W.P. Walters and B.B. Goldman Curr. Opin. Drug Discov. Devel. 8 2005
Molecular Similarity Searching Using Atom Environments, Information-Based Feature Selection, and a Naive Bayesian Classifier A. Bender, et al., J. Chem. Inf. Comput. Sci. 44 2004 10.1021/ci034207y
Binary and Numerical Descriptors¶
Descriptors can be either binary or numerical. Binary descriptors or "fingerprints", are binary bit string representations of on/off (1/0) values, each indicating the presence or absence of certain structural features in the molecule. Each bit of the fingerprint represents a descriptor, and builds a descriptor vector. Storing information in a binary presence/absence format is a computationally very efficient way to handle data. Numerical descriptors encode global or local properties of the molecule considered.

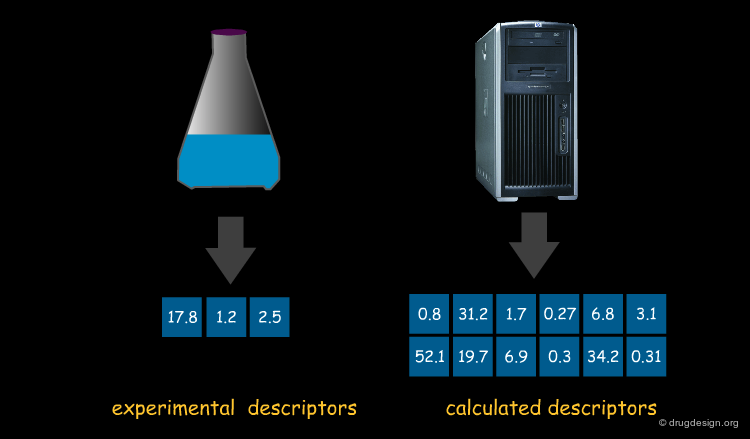
Experimental and Calculated Molecular Descriptors¶
Molecular descriptors, of which several thousand exist, can either be derived from the structure of the compound on the basis of an algorithm or they can be based on experimental data. Since experimental data are much harder to obtain than algorithms that can be applied on a structure, most descriptors are algorithmically defined.
Predefined vs. Algorithmically Defined Descriptors¶
Calculated molecular descriptors are either pre-defined or generated on the fly. This is illustrated in the figure below. One well-known class of descriptors, MDL keys, encode 166 pre-defined molecular fragments, some of which are shown below. Unity fingerprints on the other hand determine the type of atoms present between a certain number of bonds and encode whatever type of atoms are present. While MDL keys were designed to show discriminatory power between classes of compounds (and were also further optimized for this purpose), algorithmically defined descriptors are in some cases able to capture more relevant information and encode whatever structural information is given.


articles
Chemical Similarity Searching. P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
Strategic considerations in the design of a screening system for substructure searches of chemical structure files G.W. Adamson et al. J. Chem. Doc. 13 1973
Molecular Similarity Searching Using Atom Environments, Information-Based Feature Selection, and a Naive Bayesian Classifier A. Bender, et al. J. Chem. Inf. Comput. Sci. 44 2004 10.1021/ci034207y
book
Todeschini et al. Handbook of Molecular Descriptors Wiley-VCH
Possible Classification of Molecular Descriptors¶
Descriptors are often defined as one-dimensional, two-dimensional and three-dimensional descriptors. This corresponds broadly to descriptors that describe the molecule by a single value (1D) such as molecular weight, or topological indices, descriptors which employ fragments derived from the connectivity table (2D), and descriptors which capture the three-dimensional nature of the molecule (3D). All descriptors can be divided into further subtypes, but the general (although not unambiguous) classification is shown below.

Examples of Molecular Descriptors¶
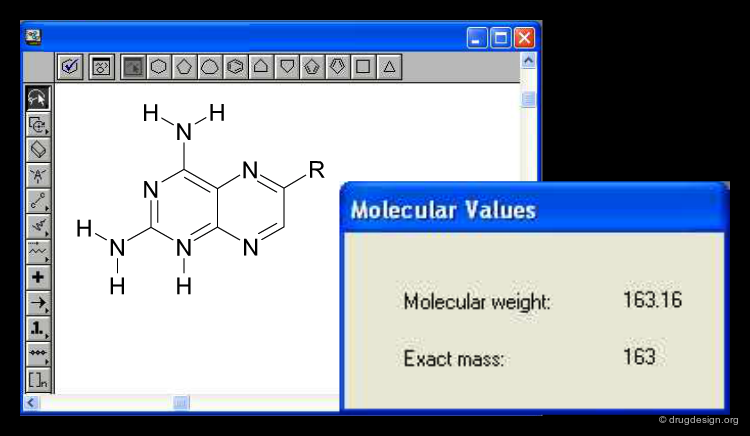
1D Descriptors: Single Numbers or Sequences¶
One-dimensional descriptors can be of two different kinds: They can either present the molecule as a single number, for example its molecular weight or its lipophilicity (logP). While this is a very short description of a molecule, it often captures a surprising degree of information relevant for classification such as for example absorption (where molecular weight is a major determinant) or narcotic effect (where lipophilicity is a major determinant of non selectively interrupting cell membranes). One-dimensional molecular representations can also be similar to the sequence of Proteins or DNA, but they are difficult to construct for 'small molecules'.
articles
The Characterization of Chemical Structures Using Molecular Properties. A Survey D.J. Livingstone. J. Chem. Inf. Comput. Sci. 40 2000
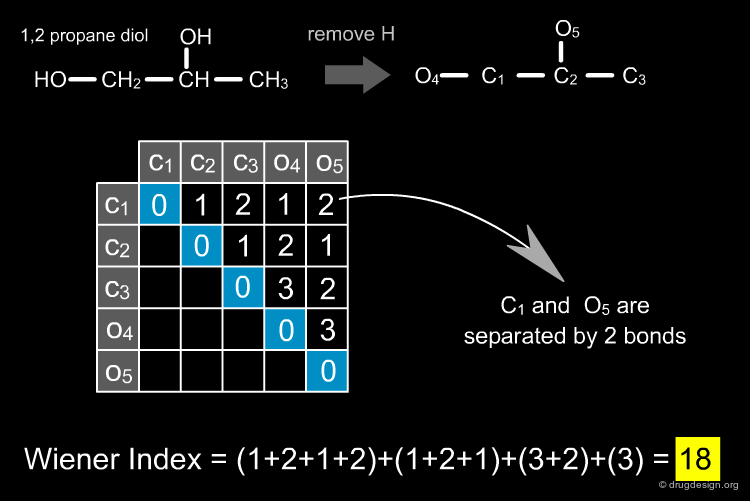
Topological Indices¶
Topological indices are derived from the connectivity table of a molecule and represent information about the topology of the structure, again captured in a single number (the "topological index"). First generation indices such as the Wiener Index are derived from integer values of the connectivity table and are themselves integers. Second generation indices are derived from integer properties but are real-valued numbers while third generation indices employ real-valued numbers for their generation and are also real-valued numbers themselves. Examples are given below. Topological indices have been widely used in QSAR studies, but their interpretation and discriminatory power remains difficult.



articles
Structural Determination of Paraffin Boiling Points H.J. Wiener J. Am. Chem. Soc. 69 1947
Recent Advances on the Role of Topological Indices in Drug Discovery Research E. Estrada and U. Uriarte. Current Med Chem. 8 2001
Electrotopological Descriptors¶
Topological indices have also been extended to describe the electronic environment of atoms in molecules. These descriptors, often known as "Kier/Hall descriptors", have proven their value in a series of applications the earliest of which was their application to the prediction of boiling points. They include both information about the electronic state of each individual atom as well as information about its environment, both of which define its characteristic properties.

articles
The electrotopological state: structure information at the atomic level for molecular graphs L.H. Hall, B. Mohney and L.B. Kier J. Chem. Inf. Comput. Sci. 31 1991
Electrotopological State Indices for Atom Types: A Novel Combination of Electronic, Topological, and Valence State Information L.H. Hall and L.B. Kier J. Chem. Inf. Comput. Sci. 35 1995
Linear Representations of Molecules¶
While DNA and proteins have "natural" one-dimensional representations due to their linear nature, this is not true for "small molecules", which may be and usually are branched and possess cyclic moieties. Nonetheless a method has been devised to represent molecules in a single dimension: this is illustrated below. Molecules are compared by aligning their one-dimensional representations and calculating the overlap of both representations as a similarity measure. Note that while in the example below the method seems to be straightforward, in practice ring systems often introduce complexity.
articles
One-Dimensional Molecular Representations and Similarity Calculations: Methodology and Validation. S.L. Dixon and K.M. Merz J. Med Chem. 44 2001
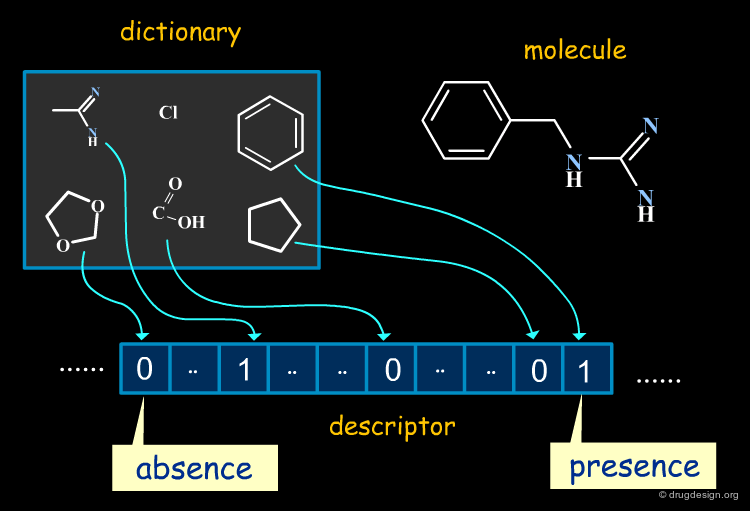
2D Descriptors: Fragments and Substructures¶
Substructural descriptors "fragment" the molecule into smaller entities and determine whether the resulting fragments match a pre-defined dictionary. They have the advantage of being easy to interpret.
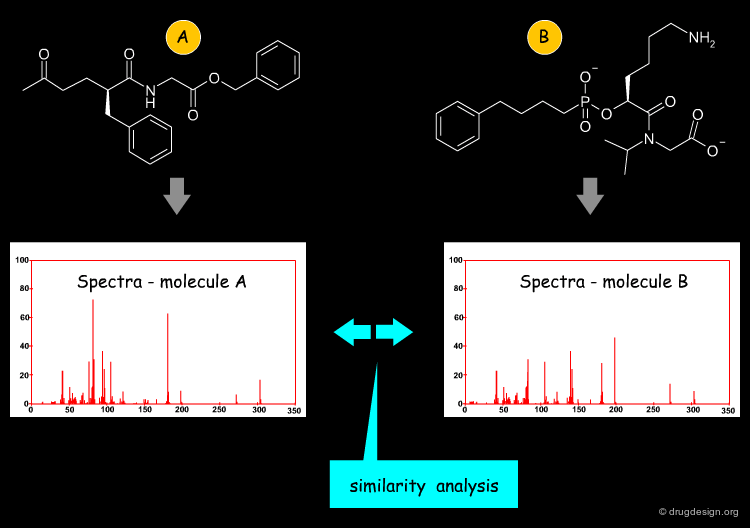
Spectra-Derived Descriptors¶
An additional method that does not calculate descriptors from the small molecule directly but uses "natural phenomena" for this purpose is employed by the class of spectra-derived descriptors. Based on either experimental or calculated spectra (which may be IR spectra or others), spectra are encoded, for example by analyzing their zero crossings. One of the methods in this category is the EVA descriptor, which is based on the vibrational absorption spectrum.
articles
The EVA spectral descriptor D.B. Turner and P. Willett. Eur. J. Med. Chem. 35 2000
Graph-Based Multiple Point Pharmacophores¶
The distance between atoms with putative interaction types can either be defined in space (as in the previous example) or it can be defined as a topological distance (i.e. by counting the number of bonds along the shortest path between two atoms). One example of this descriptor is the CATS descriptor, which employs five interaction types: lipophilic atoms, hydrogen bond donors and acceptors, and positively and negatively charged centers.

articles
'Scaffold-Hopping' by Topological Pharmacophore Search: A Contribution to Virtual Screening G. Schneider, W. Neidhart, T. Giller and G. Schmid. Angew. Chem. Int. Ed. 38 1999
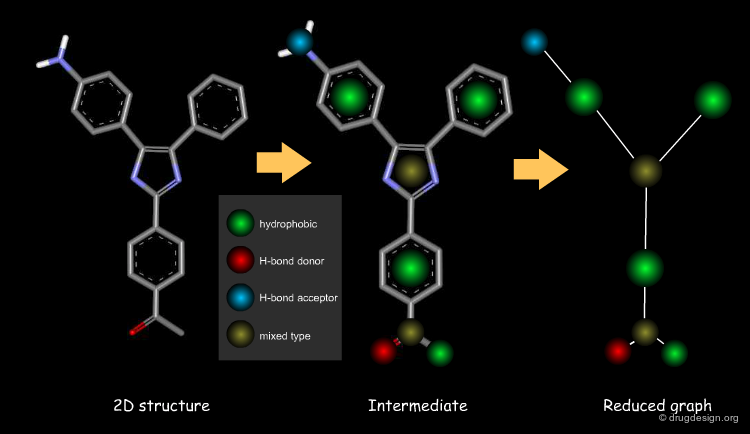
Reduced Graph and Feature Trees¶
Reduced graph and feature tree representations of molecules are similar in that they represent the molecule as a graph of edges and nodes, where nodes represent atoms (or groups of atoms) and edges represent bonds (or connections between groups of atoms). Feature trees compare two molecules by partial matching of their graph representations and the nodes include additional information about the atoms such as volume and electronic information.
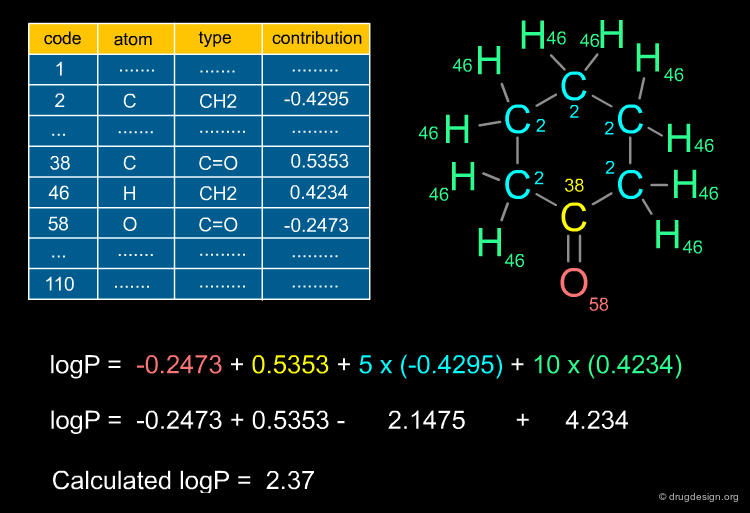
Ghose and Crippen Descriptors¶
A particular fragment definition that has gained widespread acceptance in computer-aided drug design studies was defined by Ghose and Crippen in the late 80s. By defining a set of 110 molecular features they were able to build reliable models; initially for the prediction of the octanol/water partition coefficients, but more recently this feature set has also been used for the prediction of a wide variety of molecular properties.
articles
Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships. I. Partition coefficients as a measure of hydrophobicity A.K. Ghose and G.M. Crippen J. Comput. Chem. 7 1986
. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions A.K. Ghose and G.M. Crippen J. Chem. Inf. Comput. Sci. 27 1987
3D Descriptors¶
Three-dimensional descriptors attempt to capture the 3D properties of the molecules to be compared, along with their relevant properties. A major difference between 3D descriptors and those of the 1D and 2D types, is that the three-dimensional properties are dependent on the particular conformation of the molecule. Thus, one can either use a low-energy structure of the molecule to calculate its three-dimensional descriptors, or run conformational sampling to explore the conformationally accessible space of the structure. While the second route introduces more information into the descriptor, it should be noted that at the same time it also introduces more noise, which does not necessarily improve the signal-to-noise ratio of the descriptor.

Field-Based Descriptors¶
Field-based descriptors employ molecular properties calculated in space (usually on a regular grid) to represent the molecule. The best known approach is probably Comparative Molecular Field Analysis (CoMFA), which calculates steric and electrostatic properties of molecules to define both shape and surface properties which are both important for ligand-target interactions.
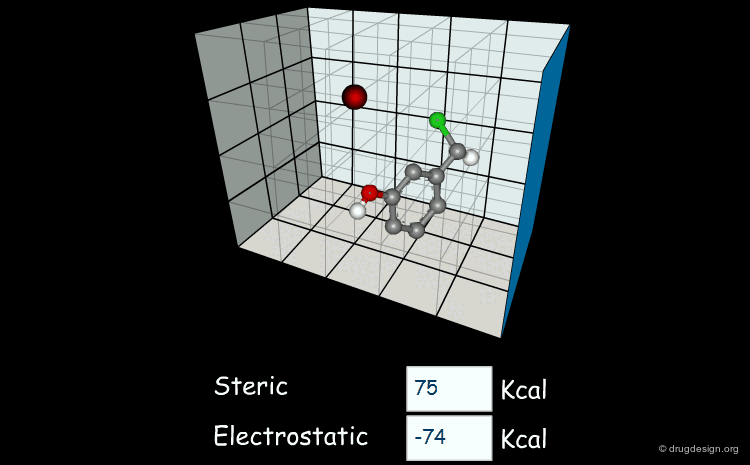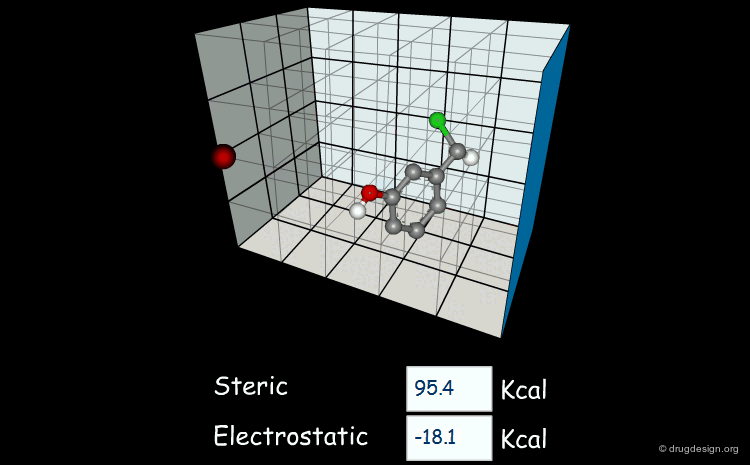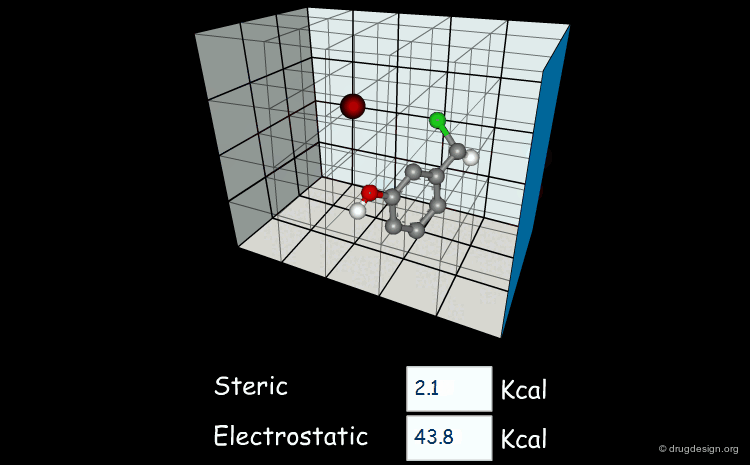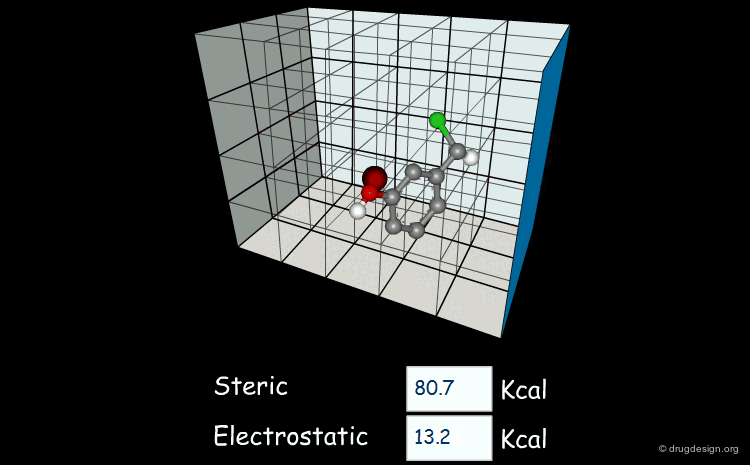
articles
Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins R.D. Cramer, D.E. Patterson and J.D. Bunce J. Am. Chem. Soc. 110 1988
A critical review of recent CoMFA applications K.H. Kim et al Perspect. Drug Discov. Des. 12 1998
Multiple-Point Pharmacophores¶
Multiple-point pharmacophores are generated in a two-step process: First, interaction types are assigned to individual atoms. Second, all pairs (two-point pharmacophores), triangles (three-point pharmacophores) or trapeziums (four-point pharmacophores) of the atom centers are constructed. All combinations of interaction types and distances can be assigned to bits in a fingerprint. First presented in the late 70s, this method has become very popular and represents one of the standard methods for virtual screening today.

articles
Three-dimensional Pharmacophore Pattern Searching P. Gund Prog. Mol. Subcell. Biol. 5 1977
3D-pharmacophores in drug discovery J. Mason. Current Pharm. Des. 7 2001
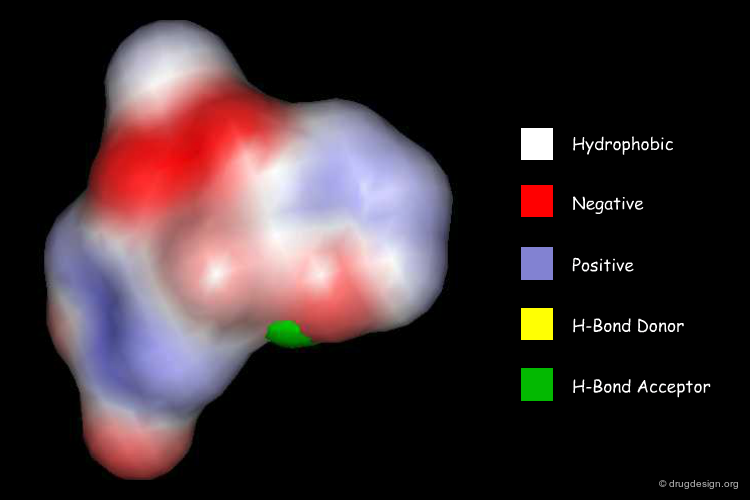
Surface-Based Descriptors¶
Surface-based descriptors do not employ points on a regular grid or the atom centers to describe a molecule: instead they use points on the molecular surface for this purpose. By doing this, encoding is simplified from a 3-dimensional problem in the case of grids to a 2-dimensional surface. Methods such as GRIND or MOLPRINT fall into this category.
articles
GRid-INdependent Descriptors (GRIND): A Novel Class of Alignment-Independent Three-Dimensional Molecular Descriptors M. Pastor, G. Cruciani, I. McLay, S. Pickett and S. Clementi. J.Med.Chem. 43 2000
. Molecular Surface Point Environments for Virtual Screening and the Elucidation of Binding Patterns (MOLPRINT 3D) A. Bender, H.Y. Mussa G.S. Gill and R.C. Glen. J. Med. Chem. 47 2004
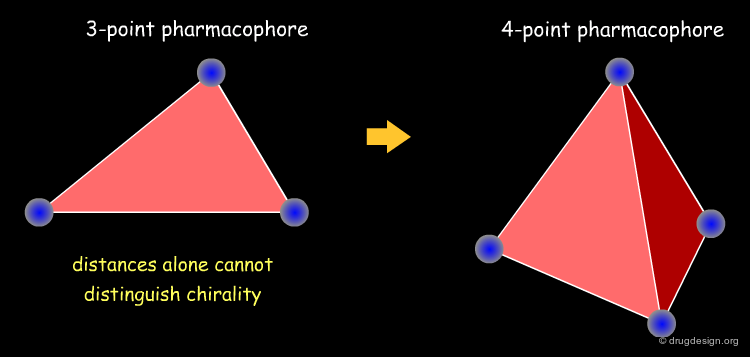
4D Chirality Descriptors¶
Chirality has often been neglected in fingerprint representations; however this has become an important issue in similarity searching. Fingerprint descriptors enabling the distinction between chiral molecules were first introduced by extending 3-point pharmacophore methods to 4-point fingerprints (Mason et al). More recently, other chirality descriptors have been proposed such as topological indices (Golbraith and Tropsha), and spectrum-like chirality codes (Aires-de-Sousa and Gasteiger).
articles
New 4-point pharmacophore method for molecular similarity and diversity applications: overview of the method and applications, including a novel approach to the design of combinatorial libraries containing privileged substructures Mason JS, Morize I, Menard PR, Cheney DL, Hulme C, Labaudiniere RF. J.Med.Chem. 42 1999
Novel chirality descriptors derived from molecular topology Golbraikh A, Bonchev D, Tropsha A. J Chem Inf Comput Sci. 41 (1) 2001
Chirality codes and molecular structure Aires-de-Sousa J, Gasteiger J, Gutman I, Vidovic D. J Chem Inf Comput Sci. 44(3) 2004
Virtual Affinity Fingerprints¶
While the methods mentioned above employ information about the ligand or inhibitor alone, virtual affinity fingerprints use an external reference frame to determine the similarity of the ligands: all structures are fitted ("docked") into a panel of diverse protein structures. For each structure, an affinity score can be calculated, resulting in a description of each compound from its affinity score vector to the protein panel. While this approach is interesting due to its external reference frame, it is also computationally very intensive.

articles
Predicting ligand binding to proteins by affinity fingerprinting L.M. Kauvar, et al. Chem. and Biol. 2 1995
Flexsim-X: A Method for the Detection of Molecules with Similar Biological Activity U.F. Lessel and H. Briem H. J. Chem. Inf. Comput. Sci. 40 2000
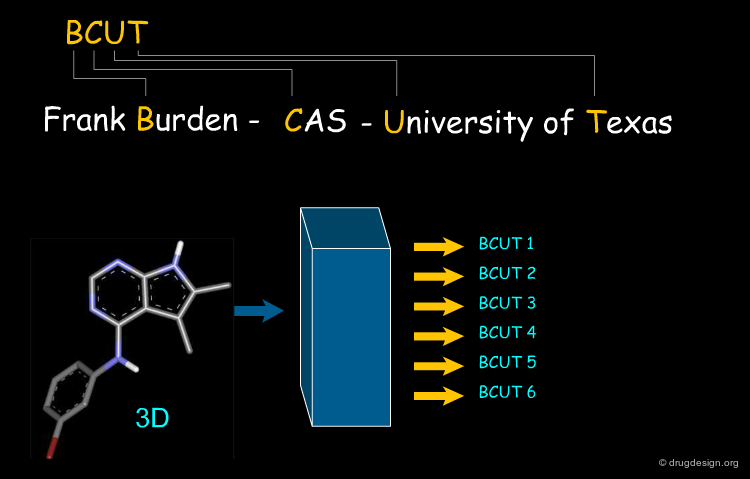
BCUT Descriptors¶
A rather abstract molecular descriptor often employed for diversity selection is the "BCUT" descriptor. It is derived from a matrix containing physicochemical properties in the diagonal elements and topological information (for example interatomic distances) in the off-diagonal elements. The BCUT descriptors are then calculated as the eigenvalues of the matrices. Its advantages are that it can be calculated quickly for large libraries and that diversity-based compound selection is easily available.
articles
Metric Validation and the Receptor-Relevant Subspace Concept R.S. Pearlman and K.M. Smith. J. Chem. Inf. Comput. Sci. 39 1999
Novel software tools for chemical diversity R. S. Pearlman and K. M. Smith Perspect. Drug Discovery Design 9 1998
Comparing Molecules: Similarity Coefficients¶
Methods to Quantify Similarity¶
In the previous section we saw how to handle molecular structures and properties in the computer by using molecular descriptors. Now we need to establish a numerical similarity measure between molecules which assigns a single number (the "similarity index") to structure pairs.
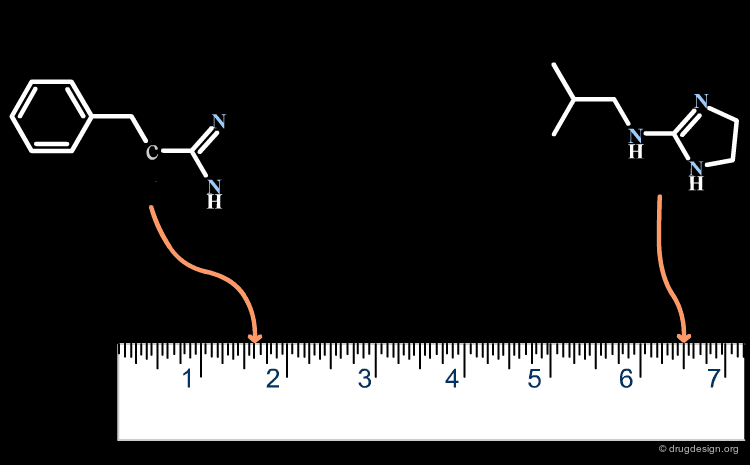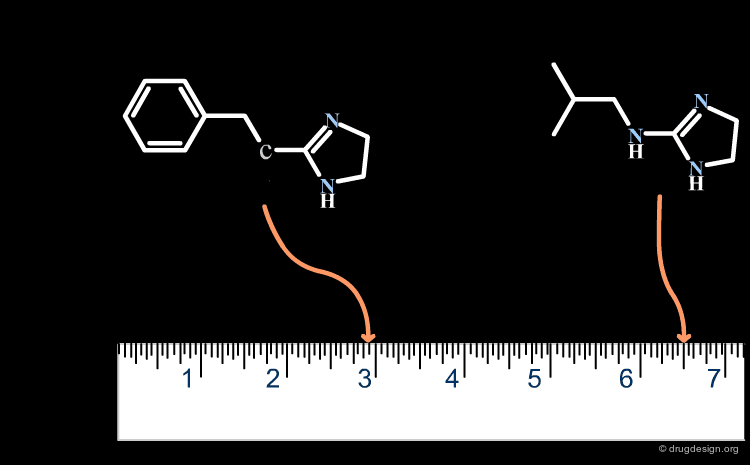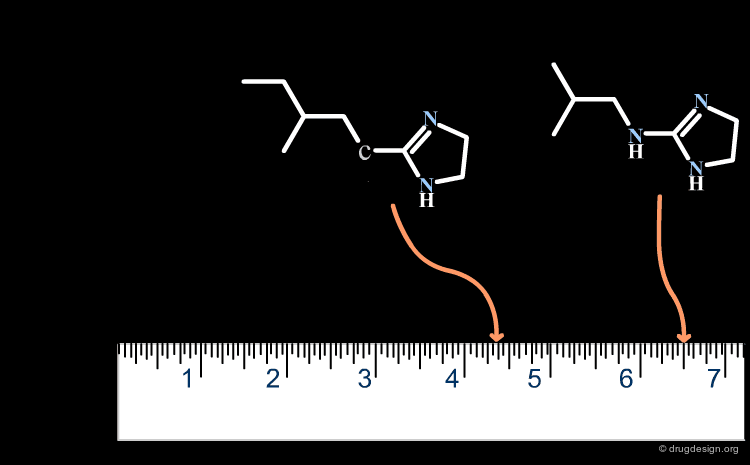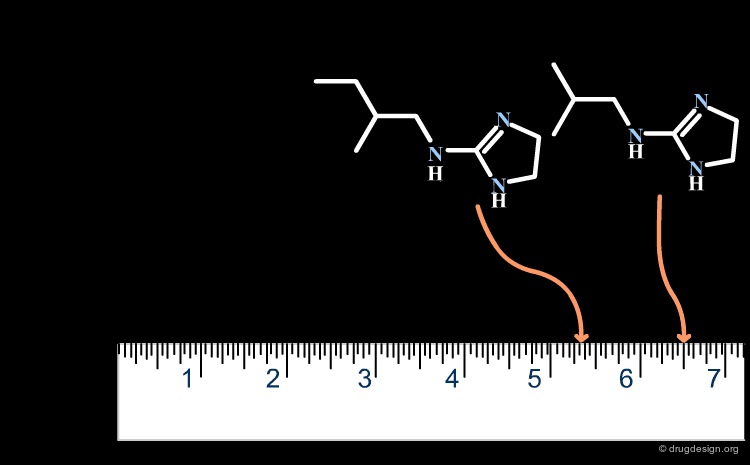
Similarity Coefficients of Relevant Properties¶
As already mentioned, similarity is a subjective concept that requires relevant descriptors that can represent the properties to be compared (e.g. biological activities). A poor choice of the relevant physicochemical properties or descriptors leads to similarity values that are meaningless.
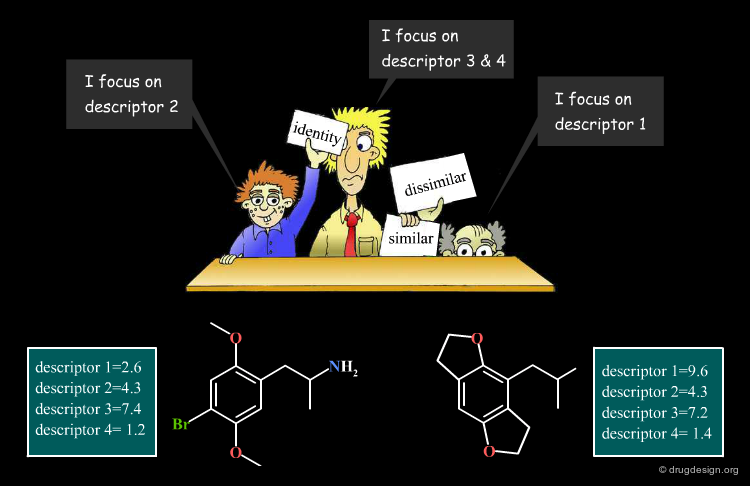
Binary and Distance-Based Formulas¶
Depending on how molecules are encoded (using either binary strings or numerical descriptors), the similarity indexes are calculated from either binary or numerical values. They are assigned the same name (e.g. Tanimoto) but have different mathematical forms. When the calculation are based on binary values the numerical similarity measure is called the "Similarity Coefficient" and when the calculations are based on numerical values it is called the "Distance Coefficient".

Distance Coefficients¶
Analogous to establishing the distance of objects in space, distance coefficients establish the distance between two descriptor representations of molecules. Distance coefficients range between 0 and infinite. One well-known distance coefficient is the Euclidean Distance, which treats the descriptor entries as dimensions in Euclidean space and is calculated as given below.

Similarity Coefficients¶
Similarity coefficients assign a value in the interval [0;1] to the similarity of molecules, with 1 representing identical items, 0 dissimilar items. Values above 0.8 are often said to designate "similar" molecules. The advantage of similarity coefficients is that their values are normalized (denominator term), and can be compared from molecule to molecule.

Symmetry Problems in Similarity Analysis¶
While similarity coefficients can be defined in many ways, one question is whether the coefficient behaves symmetrically, in other words is "A is similar to B" as "B is similar to A".

Symmetrical vs. Asymmetrical Similarity in Psychology¶
Similarity coefficients establish a numerical similarity value for a pair of items. One of the fundamental characteristics of a similarity coefficient is whether A is as similar to B as B is to A. While this seems trivial, human psychology often does not obey this principle. Using an often-cited principle, Amos Tversky asked subjects whether (a) China is more similar to North Korea or (b) North Korea is more similar to China. The second answer was given consistently; this finding is also consistent with the tendency of humans to use the larger entity (here China) as the main class and assign subtypes of the class to it. Examples of non-symmetrical indices will be presented later.

articles
Features of similarity A.Tversky Psychol. Rev. 84 1977
Does the Absence of Features Indicate Similarity?¶
Another important issue to consider is whether the absence of features should be treated as a "common property" which hints at the similarity of descriptors. This is perfectly illustrated by an example given by Sokal and Sneath: "The absence of wings among a group of distantly related organisms (such as a camel, a horse, and a nematode) would surely be an absurd indication of affinity. Yet a positive character such as the presence of wings could mislead equally for a similar heterogeneous assemblage (for example, bat, heron, and dragonfly)." For molecules, the Tanimoto index does not treat features that are not present, while distance coefficients such as the Euclidean distance do.

articles
Chemical Similarity Searching P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
book
R.R. Sokal and P.H.A. Sneath Principles of Numerical Taxonomy Freeman WH and Co 1963
Examples of Similarity and Distance Coefficients¶
In the following slides we will only discuss the most popular similarity and distance-based similarity coefficients.

The Tanimoto Coefficient¶
The Tanimoto (or Jacquard) coefficient is probably the most widely used similarity coefficient today. It is a symmetrical coefficient and provides similarities between 0 and 1. It was originally invented in 1901 to establish the similarity of populations of ecosystems that were described by the presence and absence of individual species. Due to its mathematical nature, it tends to assign higher similarity values to bigger molecules (which also usually sets more bits in the corresponding fingerprints). The binary Tanimoto coefficient is illustrated below.
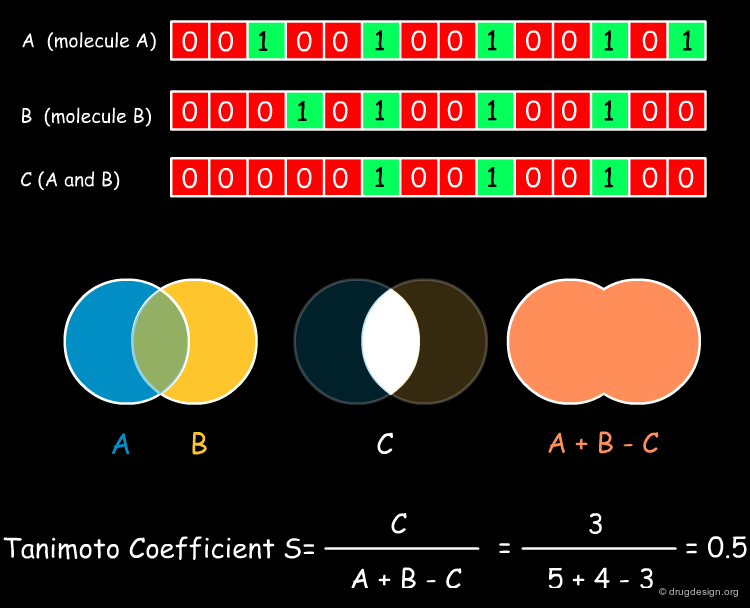
articles
Chemical Similarity Searching P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
Dice and Cosine Similarity Coefficients¶
Other popular similarity coefficients are the Dice and Cosine coefficients. They both give values in the range [0;1] and while the Dice coefficient behaves monotonously with the Tanimoto coefficient the Cosine coefficient shows high correlation with it as well. The meaning of the binary Dice and Cosine coefficients are indicated below together with that of the binary Tanimoto coefficient.
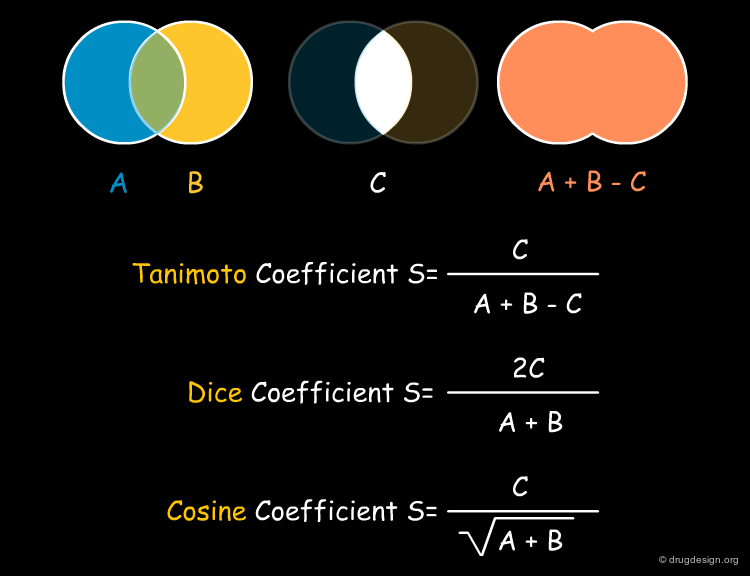
Tversky Similarity Coefficient¶
While individual similarity coefficients show either symmetrical or asymmetrical behavior, they can also be written in a more general form by incorporating free parameters. A generic form of the Tanimoto and Dice coefficients (among others) is the Tversky coefficient shown below. The free parameters α and β determine behavior of the coefficient: if they are identical, the similarity measure is symmetrical with respect to interchange of molecule A and B. In the special case of α=β=1 the Tanimoto coefficient is obtained, while in the case of α=β=1/2 we obtain the Dice coefficient.

articles
Chemical Similarity Searching P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
Features of similarity A.Tversky Psychol. Rev. 84 1977
Some Common Distance Coefficients¶
As mentioned above, Euclidean Distance is one of the most common distance coefficients employed in this area, and due to its familiarity in three dimensions it might be more accessible to human perception than other coefficients. Without taking the square root of the summed distances, the Hamming distance is obtained which behaves monotonously with Euclidean distance.

articles
Chemical Similarity Searching P. Willett. J. Chem. Inf. Comput. Sci. 38 1998 10.1021/ci9800211
Examples of Direct Use of Similarity Coefficients¶
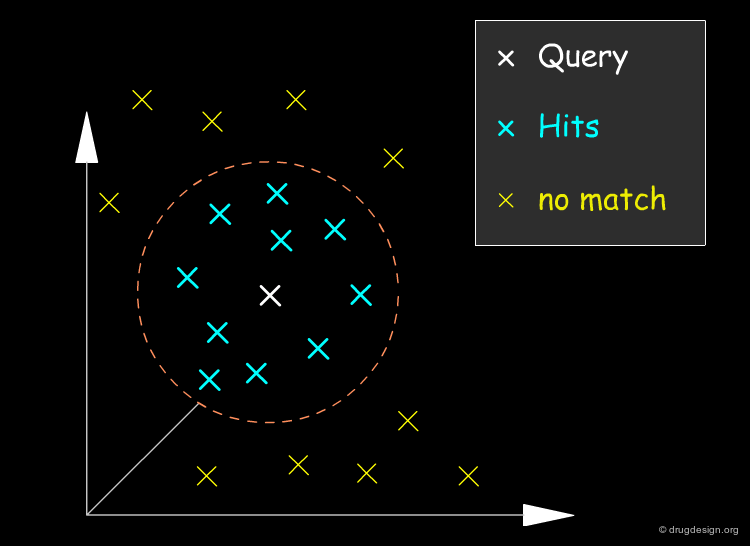
Searching Molecules with Similar Properties¶
One of the most prominent applications of the similarity approach is the task of screening a database for molecules which are similar to a known active compound. In this case, individual similarities between the query compound and the library compounds are calculated and the most similar compounds are returned to the user. For example an active compound with undesired properties (e.g. toxicity, poor solubility) can be used as a query to retrieve molecules which are similar to the original molecule in that they show the desired activity, but dissimilar with respect to the undesired properties.

Searching Information from Similar Molecules¶
Similarity searches can be used to derive information on the potential properties of a given structure. By using the structure of this compound as a query, it is possible to develop a similarity search in order to find information on the properties that were observed on similar compounds.

Knowing a Pharmacophore, Search for Novel Molecules¶
Working on a given structure having some limitations (chemical, biological or physical), it is possible to use similarity searching to find novel molecules that have a similar pharmacophore to that of the reference compound. The search will retrieve similar molecules, (possibly chemically unrelated) carrying this pharmacophore. Note that the pharmacophore query can be defined in a fuzzy way; an example of such pharmacophore is illustrated in the next page.

Example of a Fuzzy Pharmacophore¶
The following illustrates an example of a fuzzy pharmacophore. Working with TAR-RNA as a target for AIDS therapy, the authors employed a fuzzy pharmacophore to discover novel ligands of the target by similarity searching. In vitro testing of the hits obtained confirmed the predictions of the method.

articles
New inhibitors of the Tat-TAR RNA interaction found with a "fuzzy" pharmacophore model S. Renner, et al. ChemBioChem 6 2005
Validation of Novelty¶
Before embarking upon a costly synthetic program, it is important to make sure that the molecule considered is novel for the project. Has someone already thought of such a structure? Is that an original idea? Has someone synthesized this molecule? Answers to these questions can be addressed by similarity searching, which helps to assess or modify the initial idea.

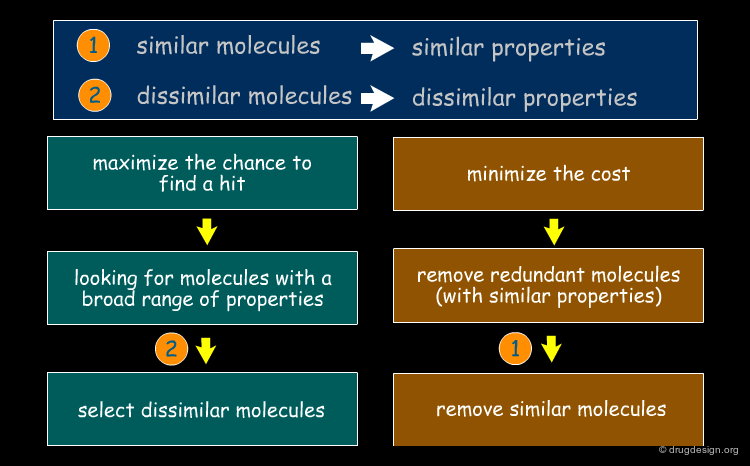
Reducing a Virtual Library to a Practical Size¶
Another application of the "molecular similarity principle" is to reduce a virtual library to a practical size of diverse molecules to be synthesized. In this case the focus is on dissimilarity rather than on similarity.

articles
Do Structurally Similar Molecules Have Similar Biological Activity? Y.C. Martin J. Med. Chem 45 2002
Diversity screening versus focussed screening in drug discovery M.J. Valler and D. Green. Drug Discovery Today 5 2000
Peptidomimetics¶
Due to their ubiquity in living systems, many natural ligands of receptors in the human body are small peptides. While it may be straightforward to apply them also directly as drugs, they have shortcomings (discussed in detail in the chapter on peptidomimetics) that make it advantageous to "hop" from peptides to non-peptide compounds. Similarity searching can be used to find small molecule compounds that carry the important structural elements of a peptide and mimic its bioactivity profile.
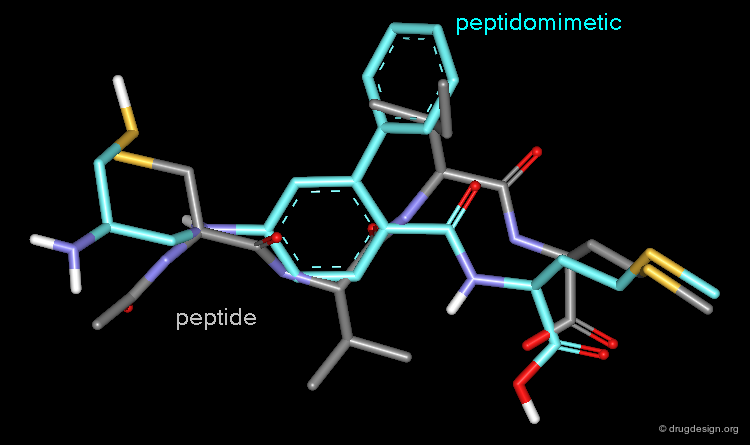
Compounds that Fit the Shape of an Active Site¶
The "molecular similarity principle" can be used for searching compounds that fit the shape of the active site of a target protein. We know that physico-chemical properties are also involved in interaction with the receptor, however similarity searching is useful for first identifying molecules that satisfy the condition of good shape complementarity.

Find a Synthetic Route¶
Similarity searching can be used for finding a synthetic route for a given molecule. It serves to identify similar molecules that were previously synthesized and adapt a particular synthetic scheme to the molecule intended to be prepared.
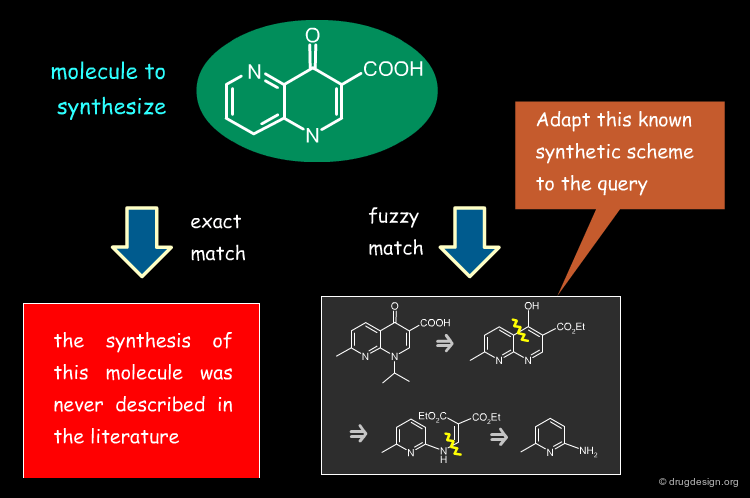
Filtering Undesired Hits¶
In drug discovery many compounds bind indiscriminately to a multitude of targets. "Ligand-based virtual screening", which is built upon the molecular similarity principle, can thus also be employed to eliminate those that are "promiscuous" - and those that are similar, from screening results. In this case the structures of promiscuous compounds are treated in a fuzzy manner by the similarity search treatments.

articles
Development of a Virtual Screening Method for Identification of "Frequent Hitters" in Compound Libraries O. Roche et al. J. Med Chem. 45 2002
Clustering of Molecules¶
Clustering is a procedure in which objects (molecules) are divided into groups. Based on the calculation of distance coefficients each compound is associated with a cluster. Clustering can be used in similarity and dissimilarity analysis, and is useful in many applications such as data mining, subset selection in library design etc...

Development of Computational Models¶
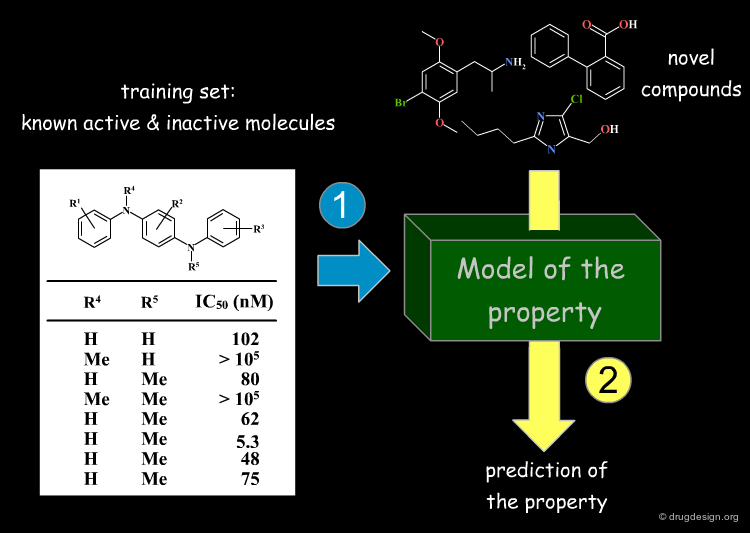
Development of a Structure-Property Model¶
Besides the direct application of searching similar/dissimilar molecules in a database, it is possible to develop a model by employing knowledge about properties of known compounds, to predict the properties of novel molecules. These models are known as "structure-property models", and are all based on the "molecular similarity principle" (a small structural change causes small property differences).

Deriving Knowledge from Distances and Properties¶
Typically, after many compounds have been synthesized and tested in a project, the medicinal chemist wants to understand SAR and identify structurally novel molecules which are similar with respect to some of their properties. The available information can be exploited to generate knowledge and understand the structure-properties relationships of a training set; this knowledge is then used to make predictions about novel molecules, based on their similarity. In the following slides we will visit the general methods these models are constructed from, followed by some typical applications.
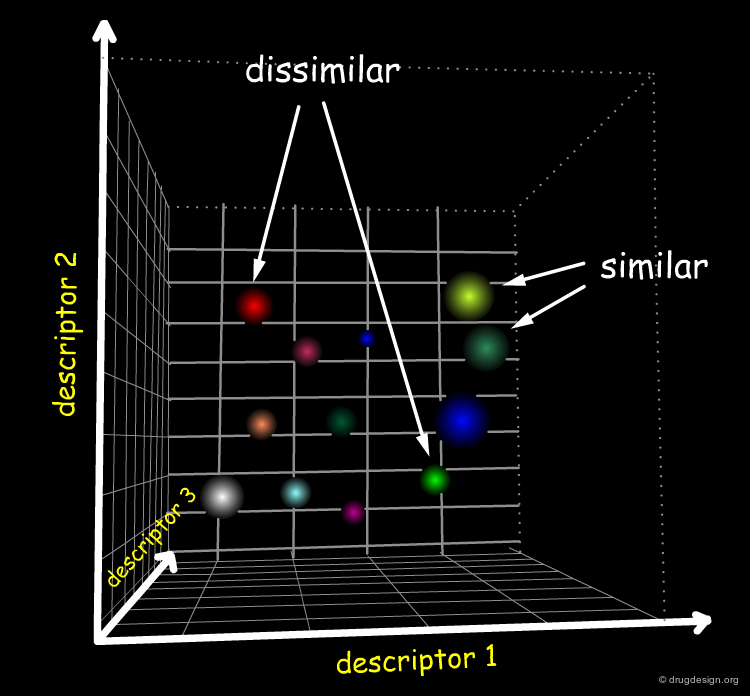
Molecules in the Space of the Descriptors¶
Contrary to the direct application of the similarity principle (where distances between molecules are explicitly calculated), in structure-property model methods, the distance between molecules are implicitly represented by molecular descriptors. These descriptors position the compounds in the space of their properties. This can be visualized by projecting the molecules in the space of the descriptors, which reveals those that are similar and dissimilar, depending on their relative location in the projection space.
QSAR and 3D-QSAR¶
QSAR (Quantitative Structure Activity Relationship) is an example of an application of the similarity principle with the use of molecular descriptors. The method is designed to formulate the relationship between structure and activity in terms of a mathematical model. For this purpose, often regression models are employed. One of the best-known methods in this area is called 3D-QSAR. This method makes it possible to compare 3D molecular forces ("fields") produced in the vicinity of different compounds to find correlations between biological activities and fields. This method has been widely employed.

QSPR - Quantitative Structure-Property Relationships¶
Apart from the prediction of activity by employing computer algorithms, drugs have to have many more desired properties which are often referred to as ADME/Tox predictions, which stands for Absorption, Distribution, Metabolism and Toxicity predictions. All of these properties can in principle be modeled by applying the molecular similarity principle.

Intelligent Machine Learning Models¶
A new trend has recently emerged, known as "machine learning", which has been triggered by high throughput developments and is aimed at providing quantitative methods to analyze screening results with their associated vast amount of numerical and non-numerical raw data, and to predict the activity of new molecules. Contrary to traditional methods that are based on numerical data, the new approaches are generally binary-based methods. Examples of such methods are indicated in the list below which is presented on the following pages.

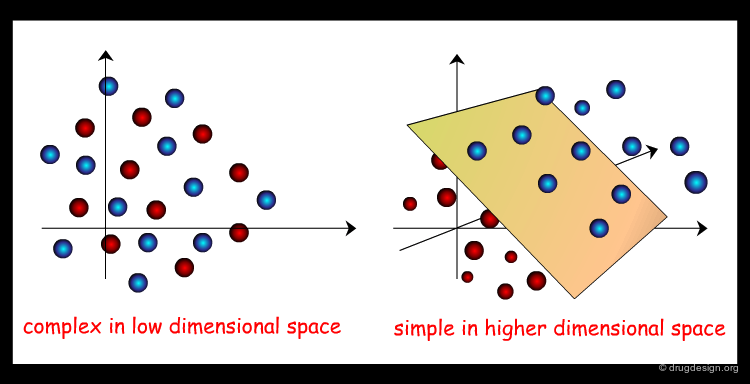
Binary Kernel Discrimination¶
Kernel Methods attempt to predict the output of a continuous output variable given continuous input variables. Kernels are used here to transform data from descriptor space into higher-dimensional space, where separation between data points can be achieved more easily. In drug-design, often only the distinction between active and non-active entities needs to be made. Binary Kernel Discrimination has been shown to perform well in similarity searching.
articles
** ** G. Harper, et al. J. Chem. Inf. Comput. Sci. 41 2001
** ** J. Hert, et al. Org. Biomol. Chem. 2 2004
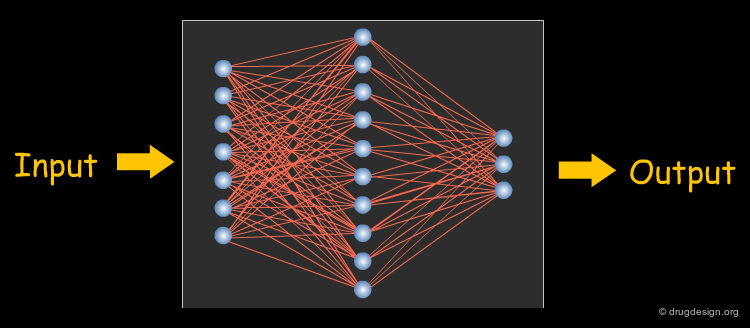
Artificial Neural Networks¶
Artificial Neural Networks (ANNs) are a supervised non-parametric classification method that can model any functional relationship between input and output variables. Thus, they are very flexible modeling tools; in fact, they are sometimes too flexible and tend to overlearn (i.e., model the training data perfectly well, but they are not able to generalize). Nonetheless, they have been very successfully applied in virtually every area of computational drug design. The architecture of a fully-connected feed-forward ANN (all nodes in each layer are connected to all nodes in the neighbouring layers) is shown in the figure below.
articles
Artificial neural networks for computer-aided molecular design G. Schneider and P. Wrede Prog. Biophys. Mol Biol. 70 1998
Support Vector Machines (SVMs)¶
Support Vector Machines (SVMs) attempt to learn the maximum separating boundary between different classes. This is a different approach than that employed by the previously mentioned neural networks that do not optimize the decision boundary if the prediction performance does not change. This different behavior is illustrated below. SVMs have been successfully applied to the discrimination between drug-like and non-drug-like compounds and also more recently to virtual screening.


articles
Active Learning with Support Vector Machines in the Drug Discovery Process M.K. Warmuth J. Chem. Inf. Comput. Sci. 43 2003
Classifying 'Drug-likeness' with Kernel-Based Learning Methods K.-R. Muller, et al. J. Chem. Inf. Model. 45 2005
Binary QSAR and Naive Bayes Classifier¶
Binary QSAR as well as the Bayes Classifier calculate the likelihood that a new compound belongs to a certain class (for example, to show bioactivity) depending on the frequency of features in active and inactive training datasets. It is very fast to train and has the advantage of being a parameter-free approach. An example of a Bayes Classifier applied to e-mail spam detection is illustrated below.

articles
Binary QSAR: a new method for the determination of quantitative structure activity relationships P. Labute. Pac Symp Biocomput.
1999
Molecular Similarity Searching Using Atom Environments, Information-Based Feature Selection, and a Naive Bayesian Classifier A. Bender, et al. J. Chem. Inf. Comput. Sci. 44 2004 10.1021/ci034207y
Rule-Based Approaches¶
Rule-based learning means inferring human-understandable rules for a certain behavior from data deductively. Below the rules determining activity of inhibitors binding to a protein kinase are given. Not every relationship between input data and output data can be modeled this way, but interpretability is often a major advantage of this method.

articles
New Gener. Comput. R.D. King et al
13 1995
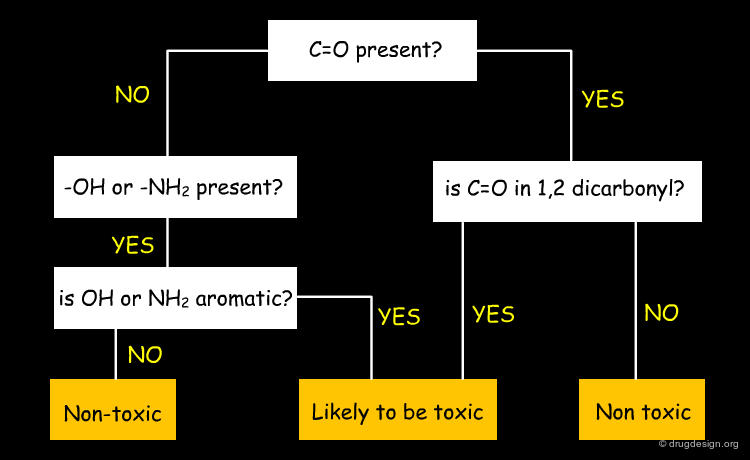
Decision Trees¶
Decision Trees form a hierarchical tree of yes/no decisions to determine class membership of the data. Decisions having the best explanatory power should be made first, in order to explain the individual parts of a dataset with the smallest number of decisions possible. Below a (hypothetical, but realistic) example from the prediction of toxicity is given. Again, decision trees have the major advantage of being easy to interpret.
Practical Applications of Structure-Property Models¶
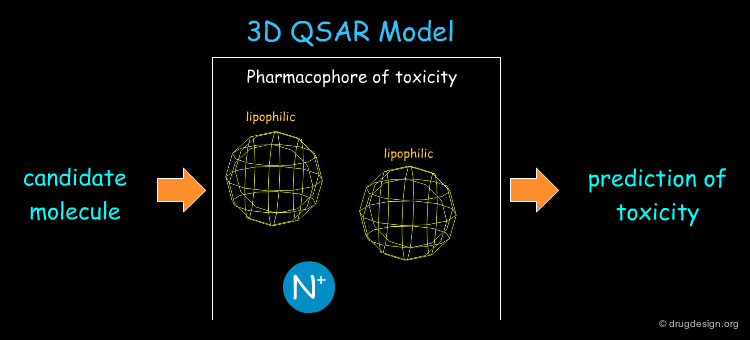
Example of a 3D-QSAR Model¶
The hERG potassium channel is involved in the polarization and repolarization of the heart muscle with every hearbeat. Unfortunately, deaths occurred since drugs on the market blocked this channel, leading to arrhythmias of the heart and finally to cardiac arrest. A 3D-QSAR model derived from a set of compounds for the hERG channel consists of a positively charged nitrogen and two lipophilic moieties and is shown below. This knowledge is crucial to avoiding the design of toxic compounds.
articles
Toward a Pharmacophore for Drugs Inducing the Long QT Syndrome: Insights from a CoMFA Study of HERG K+ Channel Blockers Andrea Cavalli J. Med. Chem. 45 2002
Molecular Similarity: Models of ADME/Tox Predictions¶
Besides bioactivity, pharmaceutical compounds also have other criteria to fulfill, such as being non-toxic to the body. Often many of these additional criteria are referred to as "ADME/Tox" properties, which are explained below. A=Absorption. How well is the compound absorbed (=able to enter the bloodstream) if taken orally? D=Distribution. Is the compound enriched in undesired 'compartments' of the body or does it reach the relevant parts of the body? M=Metabolism. Is the compound converted in the body, and if, to which metabolites and how fast? E=Excretion. For how long does the compound remain in the body and which routes does it take to leave it? Tox=Toxicity. Is this compound toxic, meaning that it has other bioactivities besides the desired one?

'A' - Absorption: Does a Drug Work Orally?¶
Absorption in the gut is achieved via different mechanisms, including passive diffusion and active transport. Still, most often passive transport occurs and for this simple case a good correlation for example with the molecular weight or the size of the compound can be established, based on the molecular similarity principle.

'D' - Distribution: Where Does the Drug Go in the Body?¶
Distribution of compounds can be modeled using a "compartment-system" of the body, representing bloodstream, fatty tissue compartment, water compartment etc. Based on the assumption that similar molecules show similar distribution in tissues, distribution models can be built. One property of relevance here is logP, which is logical since it defines the likelihood that a given compound is able to cross membranes.

'M' - Metabolism: The Drug's Fate¶
Metabolism prediction can be broadly based on an understanding of underlying mechanisms or the statistical analysis of a large data base. One recently published statistical method employs a metabolism database (the MDR Metabolite Database) to analyze the type of substructures that are most often attacked in humans or other living beings and converted to degradation products. The circular fingerprints employed, which determine substructural similarity (and thus the likelihood that the compound will undergo the same reaction, based on the scoring of metabolic sites) are shown below.
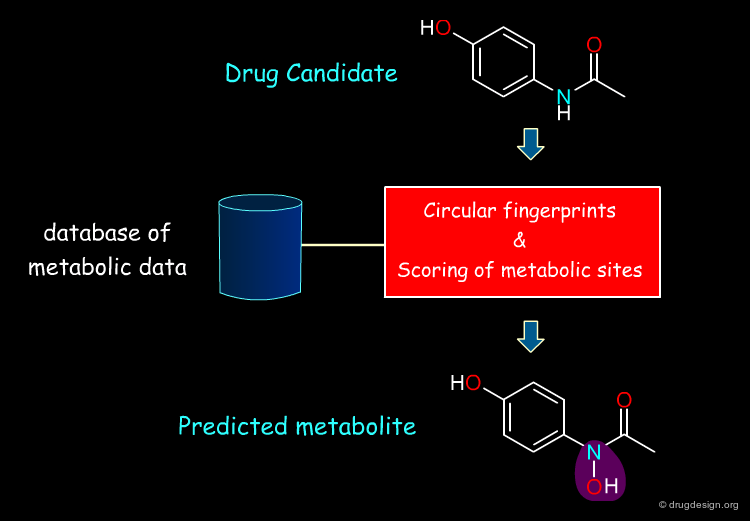
articles
SPORCalc - Fingerprint-based Probabilistic Scoring of Metabolic Sites Hasselgren-Arnby, et al. J. Chem. Inf. Model.
2005
Prediction of Putative Reaction Centres in Xenobiotics from Metabolite using Substructural Fingerprinting Smith, J. et al. Biochemical Society Transactions 31 2003
'E' - Elimination: The Drug Says Good-Bye¶
The elimination of compounds is closely linked to their lipophilicity, or in turn, to their solubility in water. Thus the molecular similarity principle works well in this area, and a correlation between the rate of elimination and logP of a series of compounds is shown below. Note that the very lipophilic compounds undergo increased metabolism and conjugation to hydrophilic moieties, so the original lipophilicity of a compound is only partly responsible for its speed of elimination.

'Tox' - Toxicity: Side-Effects¶
One well-known system for the prediction of toxicity is DEREK. It is rule-based and performs exact substructural matching for the determination of possible toxic effects. Some of its learned rules are illustrated below.

Prediction of Solubility¶
Physicochemical properties such as solubility are often easier to predict than properties such as bioactivity since they are non-directional (one does not have to consider external forces such as the receptor, which might for example cause steric repulsion). Solubility is determined by interactions between the molecular surface and the solvent. Strong correlations can be seen for example between the dipole moment or the charge or the lipophilicity of a molecule and its solubility. Still, interactions in the solid state also influence solubility and these are difficult to predict.

Prediction of Melting Points¶
Melting points are determined by the interactions of compounds in the crystal lattice. Since crystal structures can change abruptly even in the event of comparably small changes in molecular structure it is difficult to apply the molecular similarity principle based on knowing the monomer small molecule alone. Nevertheless, correlations can be seen between the rigidity of the structure and the melting point.

articles
Molecular Descriptors Influencing Melting Point and Their Role in Classification of Solid Drugs C.A. Bergstrom et al. J. Chem. Inf. Comput. Sci. 43 2003
General Melting Point Prediction Based on a Diverse Compound Data Set and Artificial Neural Networks M. Karthikeyan, Robert C. Glen and A. Bender J. Chem. Inf. Model. 45 2005
Important Properties of Molecular Descriptors and Similarity Coefficients¶
Neighborhood Behavior¶
If descriptors are employed for similarity searching, the goal is to determine property similarity, with descriptor similarity being the practical means towards this goal. Thus, correlation between the distance in property space (what one is really interested in) and in descriptor space (what one is able to determine computationally) is highly relevant. The concept of "Neighborhood Behavior" describes high correlations between descriptor similarity and property similarity which is crucial for practically every relevant molecular descriptor.

articles
Neighborhood Behavior: A Useful Concept for Validation of "Molecular Diversity" Descriptors D. E Patterson, et al. J. Med. Chem. 39 1996
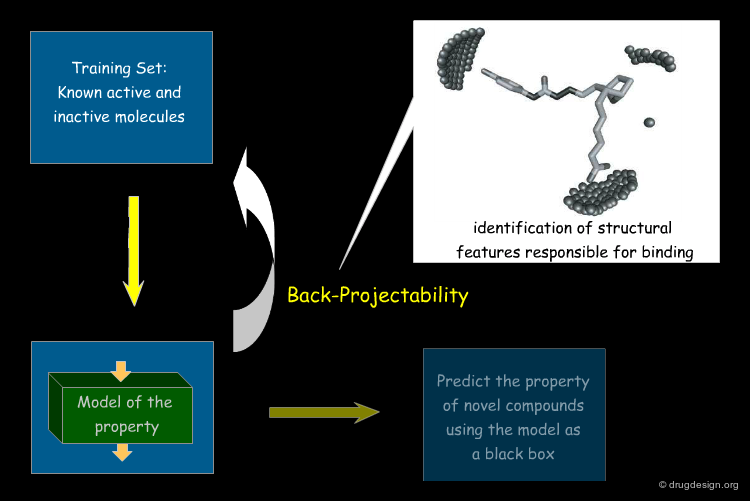
Back-Projectability¶
One aim of property models is to make sensible predictions about novel compounds. Of additional interest is the following question: which structural features actually make this particular compound so active, soluble or toxic? Back-projectable descriptors try to answer this question by projecting features found to be important back onto the molecular structure: they are back-projectable.
articles
Mapping Property Distributions of Molecular Surfaces: Algorithm and Evaluation of a Novel 3D Quantitative Structure-Activity Relationship Technique N. Stiefl and K. Baumann J. Med. Chem. 46 2003
Molecular Surface Point Environments for Virtual Screening and the Elucidation of Binding Patterns (MOLPRINT 3D) A. Bender et al. J. Med. Chem. 47 2004
Validation and Information Content of Descriptors¶
While a multitude of molecular descriptors is available, one should make sure that they actually capture relevant information and are able to reliably make predictions. For virtual screening and also QSAR as well there are several standard datasets available. But do not overestimate the information content of seemingly sophisticated descriptors, since performance depends enormously on the property of interest and the particular class of molecules. Recently it has been shown that even today's best descriptors do not perform much better than if one just counts atoms on one of the benchmark virtual screening datasets.

articles
Discussion of Measures of Enrichment in Virtual Screening: Comparing the Information Content of Descriptors with Increasing Levels of Sophistication. Andreas Bender and Robert C. Glen, A J. Chem. Inf. Model. 45 2005
Properties of Binary Fingerprints¶
Due to their binary nature, the distribution of similarity coefficients usually does not tend to be random. Simple ratios such as 1/2, 1/3 and 2/3, etc. are much more frequent than other ratios with larger numerators and denominators. For small fingerprints it was shown that 1/3 is the most frequent similarity value for an infinitely long bit string which is half occupied, a value that should be kept in mind to have a feeling for similarity values.

articles
Combinatorial Preferences Affect Molecular Similarity/Diversity Calculations Using Binary Fingerprints and Tanimoto Coefficients J.W. Godden, L. Xue and J. Bajorath. J. Chem. Inf. Comput Sci. 40 2000
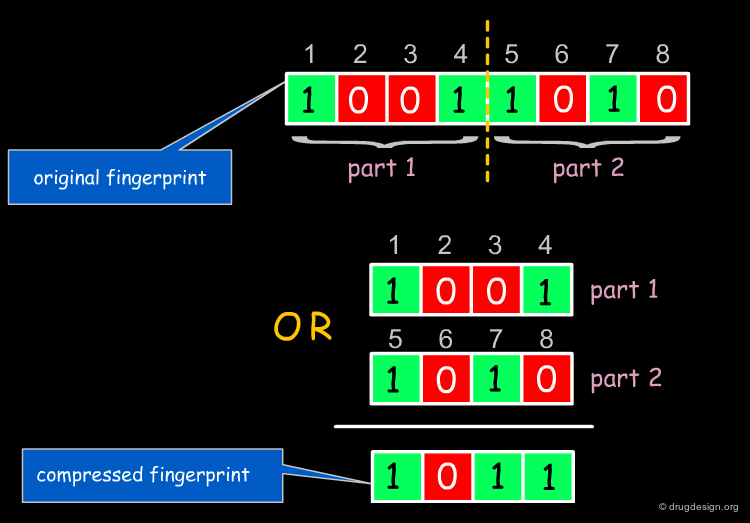
Folding of Fingerprints¶
It is sometimes useful to compress the information encoded in long fingerprints. In this case, the whole fingerprint is said to be "folded". It is cut into two halves, which are combined by a logical OR (or other) operation to give a new fingerprint of only half the size of the original fingerprint. In the resulting fingerprint, bits are set at position n if they were present in position n of the original fingerprint OR position (length/2)+n (which corresponds to bit n of the "second half" of the fingerprint). This process can also be repeated.
The Concept of Binning¶
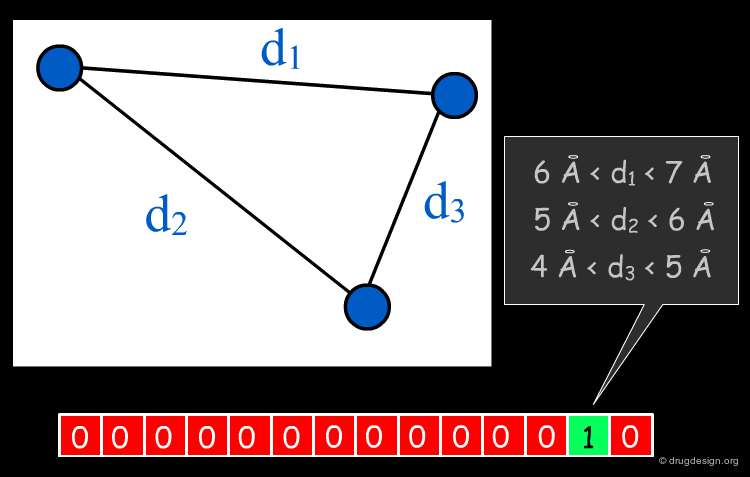
If descriptors of molecules involve the distance of intermolecular features in space and one wants to determine whether identical (or equivalent) features are present in two molecules, exact matching of a particular distance virtually never occurs. In order to still be able to compare spatial arrangements of features a concept called "binning" is often employed, where ranges of intra-feature distances are assigned to the same "distance range", also called a "bin".
The Concept of "Fuzzy" Descriptors¶
If distances between features are binned, what means that they are assigned to a discrete distance range (for example according to the rule "a distance between 6 and 7 Angstroms corresponds to bin 7"), very similar distances might still be assigned to different bins. This happens around the bin borders; in the example above a distance of 5.9 Angstroms would be assigned to bin 6, but a (very similar) distance of 6.1 Angstroms would be assigned to bin 7. "Fuzzy" descriptors also increase the counts of neighboring bins, thereby alleviating the problem of discretizing real-valued distances. An example is given in the applications section.

articles
New inhibitors of the Tat-TAR RNA interaction found with a "fuzzy" pharmacophore model S. Renner, et al. ChemBioChem 6 2005
Size-Bias of the Tanimoto Similarity Coefficient¶
Both the molecular representation in descriptor space and the comparison via similarity coefficients emphasize certain molecular features or properties while they neglect others. One of the most important properties of the Tanimoto coefficient is its size-bias as illustrated in the biphenyl query shown below. The Tanimoto coefficient reaches a limit and does not decrease when the size of the second molecule is increased.

articles
On the Properties of Bit String-Based Measures of Chemical Similarity D.R. Flower J. Chem. Inf. Comput. Sci. 38 1998
Size-Bias: Favoring Large Molecules¶
Similarity coefficients can usually not be compared directly; the size of the query has a major influence on the distribution of similarities. In the following figure is illustrated the distributions of Tanimoto coefficients between a query and all molecules of a database. The larger the query, the higher the average similarity, and the wider the distribution of values. This is also known as a 'size-bias' of the Tanimoto coefficient: in similarity calculations larger compounds are favored.

articles
On the Properties of Bit String-Based Measures of Chemical Similarity D.R. Flower J. Chem. Inf. Comput. Sci. 38 1998
2D vs. 3D Descriptors¶
Two-dimensional and three-dimensional descriptors also have specific advantages and disadvantages. While each descriptor also shows individual differences, the following general rules can be given: 2D descriptors perform well where invariant topological features constitute a given activity, they perform less well in the case of topologically more diverse molecules which all show the same property. 3D descriptors on the other hand are able to identify structures with different topologies that still show the same activity, a capability often referred to as "scaffold hopping". On the other hand, they are slower and often found to be less efficient for example in virtual screening runs.


Choice of the Best Method for Calculating Similarity Coefficients¶
Unique Content of Each Similarity Coefficient?¶
While many similarity coefficients can be defined, and indeed have been, the question arises as to whether they contain different information from each other or whether they behave similarly; in other words, whether they cluster into coefficients with very similar properties. While the first step is to investigate whether or not similarity coefficients show similar behavior, we might also ask a second question: if similarity coefficients indeed show different properties, might it be advantageous to combine them? Both questions will be explored in the following pages.

Clustering Similarity Coefficients¶
In a recent work 22 similarity coefficients were analyzed with respect to their behavior, and 3 distinct clusters could be identified. This may suggest that many of them were redundant. A graphic representation of the correlation between descriptors is visualized below (the smaller the distance, the higher the correlation between the descriptors).


articles
Grouping of Coefficients for the Calculation of Inter-Molecular Similarity and Dissimilarity using 2D Fragment Bit-Strings J. D. Holliday, C.Y.Hu and Pl. Willett. Comb. Chem. HTS 5 2002
Consensus Scoring: Asking a Panel of Experts¶
Imagine you are on a TV quiz show and have to answer a set of questions ranging from any area of sports to any area of culture. You have two people who are allowed to help you; one is better at sports (and okay at culture), one is better at culture (and okay at sports). But you don't know who's better in which area since the persons are anonymous. Whom do you trust? One option is to ask both people and take both opinions into account. This is precisely the concept of "consensus scoring". This concept can be applied in Similarity Analysis.

articles
Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. Charifson, P. S.; Corkery, J. J.; Murcko, M. A.; Walters, W. P. J. Med. Chem. 42 1999
Consensus Scoring Criteria for Improving Enrichment in Virtual Screening. Yang, J. M.; Chen, Y. F.; Shen, T. W.; Kristal, B. S.; Hsu, D. F. J. Chem. Inf. Model. 45 2005
Why Does Consensus Scoring Improve the Results?¶
Mathematically speaking, consensus scoring improves the results in cases where individual errors are independent of each other: they cancel out. This is not true in every case, but on average better results are obtained. On the other hand, if the predictions errors are not (at least partly) independent of each other, consensus scoring is unable to improve results.


articles
Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. Charifson, P. S.; Corkery, J. J.; Murcko, M. A.; Walters, W. P. J. Med. Chem. 42 1999
Consensus Scoring Criteria for Improving Enrichment in Virtual Screening. Yang, J. M.; Chen, Y. F.; Shen, T. W.; Kristal, B. S.; Hsu, D. F. J. Chem. Inf. Model. 45 2005
What Algorithms Exist for Consensus Scoring?¶
One can merge the predictions from multiple classifiers in many ways: One can use the sum of the individual predictions, the relative ranks or the highest prediction. In practice, the sum of individual predictions has been found to perform best in some cases. In others the highest score assigned to each individual prediction performed best. It should be noted that no generally applicable rules exist here unfortunately.

articles
Combination of molecular similarity measures using data fusion C.M.R. Ginn, P. Willett and J. Bradshaw. Perspect. Drug. Discov. Des. 20 2000
Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures J. Hert, et al. Org. Biomol. Chem. 2 2004
Limitations of the Concept of "Molecular Similarity"¶
Limitation of Ligand-Based Approaches¶
The "molecular similarity principle" is generally used when the biological receptor (or the mechanism of action) is not known, and this lack of knowledge represents the most fundamental limitation (but also its strength!) of all ligand-based approaches: when focusing only on the ligands, everything cannot be explained. Ignoring the receptor introduces a bias with the risk of incorrect interpretations. This is discussed in the following pages.

Example of Ligand-Based Approach Limitation¶
Below, two very similar structures are shown Caffeine differs from Theophyllin purely by a methyl group, a very small change overall. Still, RNA binding is decreased by a factor of 10,000, which would not be expected based on the molecular similarity principle. If we ignore the interactions with the receptor, we cannot explain this difference.
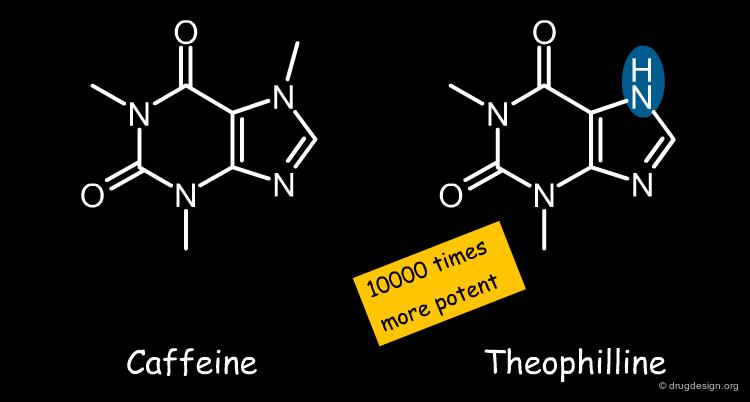
articles
Interlocking structural motifs mediate molecular discrimination by a theophylline-binding RNA G. R. Zimmermann, et al. Nat. Struct. Biol. 4 1997
Limitation Due to Extrapolations¶
The first limitation of the principle of similarity concerns the distribution of the molecules of the initial dataset. While it may straightforward to extrapolate the properties of closely related objects (e.g. A or B), the extrapolation for C becomes highly debatable. The only way to overcome this limitation is to cover a large range of values for those descriptors believed to be relevant to the property at hand.

Limitation Due to Pitfalls in interpolations¶
A second type of limitation that is more difficult to predict concerns the possibility of pitfalls in interpolations. Despite the relatively close proximity of A to a set of similar objects, the prediction of its properties may not be straightforward for reasons explained in the following pages.

Principle of Continuity¶
Because of our ignorance of the interactions with the receptor, the molecular similarity principle (based on ligand-based methods) relies on the principle of "continuity", assuming that changes are gradual in the recognition of molecules. Therefore, the similarity principle is not applicable when abrupt changes occur in the system.
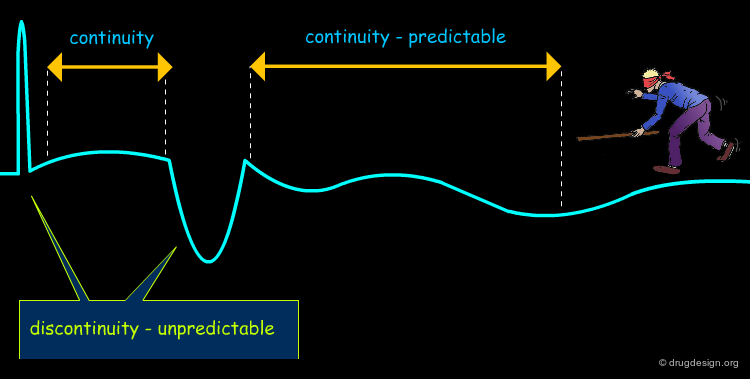
Discontinuity in Molecular Recognition¶
Examples of cases where abrupt changes occur and the similarity principle is not applicable include the following: bumps, ligand or receptor conformational changes, flip in binding modes, discontinuity in ligand properties or in receptor function.
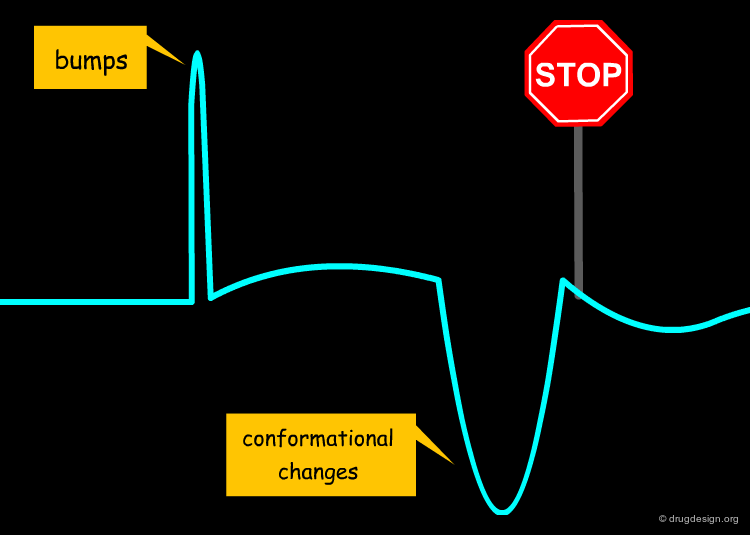
Bumps¶
A small change in the structure of a ligand can result in dramatic changes of the biological properties. For example in the structure below, the simple move of one nitrogen atom in the pyrimidine ring transforms a potent inhibitor into an inactive molecule. This may be due to bumps with the receptor, a discontinuity in the activity that in the first place, is not foreseeable (see button "Explanation"). Here, the similarity principle is not applicable.
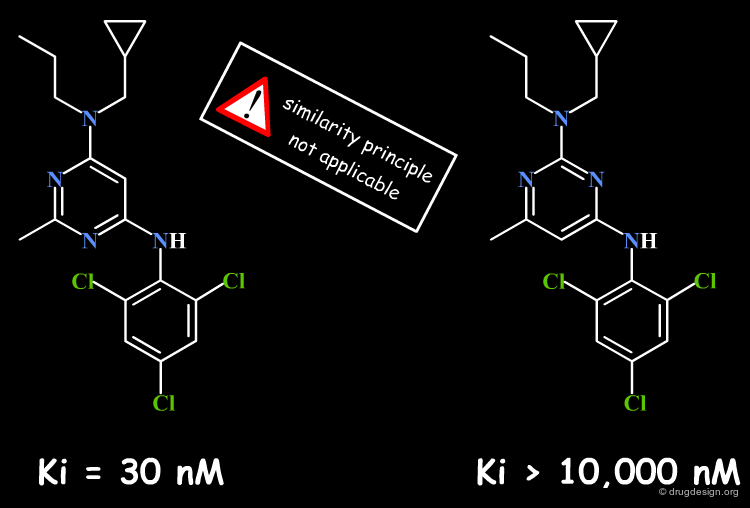

articles
Design and Synthesis of a Series of Non-Peptide High-Affinity Human Corticotropin-Releasing Factor1 Receptor Antagonists C. Chen, et al. J. Med. Chem. 39 1996
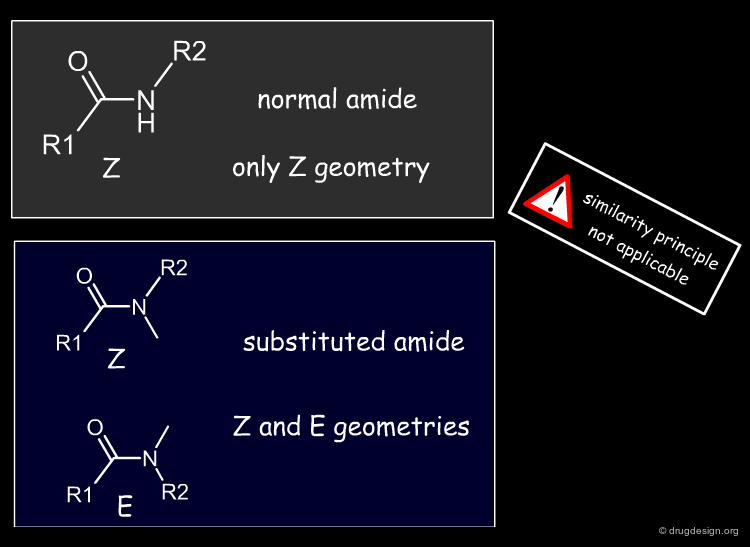
Ligand Conformational Change¶
A change in the structure of a ligand can result in an important change in the conformation of the molecule, and the principle of continuity cannot be invoked in such cases. Examples of conformational changes are shown below, where only one carbon atom is added to the initial structure. The similarity principle is not applicable.

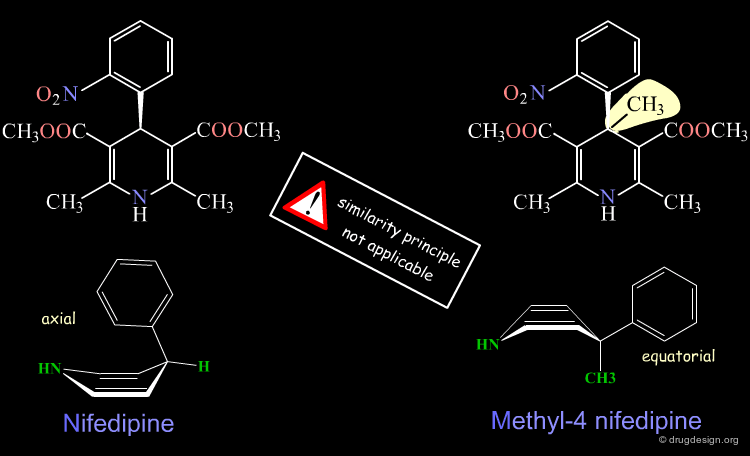
Receptor Conformational Changes¶
For reasons that are not predictable, the receptor may undergo substantial conformational changes. In the example below the enzyme renin adopts different conformations (blue and red) depending on the inhibitors. Note the change of a key tyrosine residue, which exhibits favorable interactions with the inhibitor (yellow); while with the red conformation it clashes. The similarity principle is not applicable when this is overlooked.

Flip in Binding Mode¶
When an X-ray structure of a complex is available for a given ligand, the design normally includes the assumption that the binding mode of the analog will be similar to that of the reference molecule. This has proven to be valid in many cases. However, there are examples where important flips were observed (in some cases a 180 degree rotation of the structure), and this is another limitation of the above mentioned continuity principle. The similarity principle is not applicable.
Discontinuity in Ligand Property¶
A small alteration of a structure may introduce substantial changes in specific physico-chemical properties that are not apparent in the first place. For example, the replacement of a carbon atom by an oxygen, or the change in the number of carbon atoms in a molecule can affect many properties of the substance. Examples of changes in pKa and logP are presented on the following pages.

pKa¶
The change of the subtituents in the cocaine structure results in important changes in the pKa values with repercussions on biological activities. In this example the non-continuity that occurred in the pKa values were well exploited, but in other situations such considerable changes might be overlooked.
articles
Conformational, Aqueous Solvation, and pKa Contributions to the Binding and Activity of Cocaine, WIN 35 065-2, and the WIN Vinyl Analog. Yang B, Wright J, Eldefrawi ME, Pou S and MacKerell AD HJ. Am. Chem. Soc 116 1994
Chemistry, Design, and Structure-Activity Relationship of Cocaine Antagonists Singh S Chem. Rev 100 2000
LogP¶
When changes are introduced in a structure, their impact on the global characteristics of the modified compounds must be taken into account. The example below illustrates a change in logP that is not negligible.
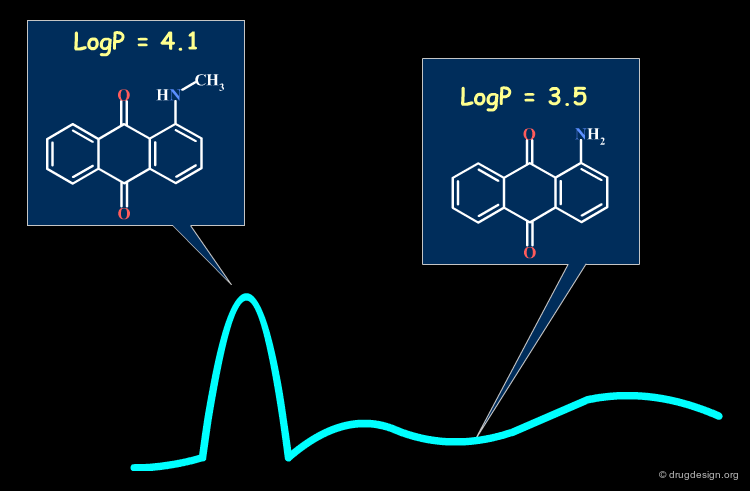
Discontinuity in the Function of the Receptor¶
A small change of a ligand can result in dramatic alteration of effect with a receptor. For example L 162,389 and L 162,782 differ only by one methyl group. The former is an antagonist and the latter an agonist of the angiotensin AT1 receptor. Appreciate the dramatic change in receptor binding that occurs for all the examples shown below. A conformational change of the receptor is initiated by the agonist but not the antagonist: a discontinuity that is very hard to predict.



articles
Benzodiazepine Binding Site Qinmi Wang, Yifan Han, and Hong Xue CNS Drug Reviews Vol. 5, No. 2 1999
Dual Agonistic and Antagonistic Property of Nonpeptide Angiotensin AT1 Ligands: Susceptibility to Receptor Mutations Signe Perlman, Claudio M. Costa-Neto, Ayumi A. Miyakawa, Hans T. Schambye, Siv A. Hjorth, Antonio C. M. Paiva, Ralph A. Rivero, William J. Greenlee, and Thue W. Schwartz Molecular Pharmacology 51 1997
Conclusions¶
How Does "Molecular Similarity" Fare Today?¶
Despite of the limitations of ligand-based design, "Molecular Similarity" is a concept routinely applied in pharmaceutical and related industries today. Molecular similarity methods are very much of importance in the early stages of drug discovery. They are most often applied when little knowledge is available (e.g. only one active compound). With increasing knowledge, their importance generally decreases and vanishes when the structure-based level is reached.
How Many Chances of Being Active?¶
In an attempt to answer the question "do structurally similar molecules have similar biological activity?" a follow-up study to 115 high-throughput screening assays shows that at high Tanimoto similarity values (>0.85), only 30% of compounds similar to an active molecule are themselves active. This reveals the complexity of molecular similarity. A list of possible factors explaining such observations is given below.

articles
Do Structurally Similar Molecules Have Similar Biological Activity? Y.C. Martin J. Med. Chem 45 2002
Economic Rationale of Similarity-Based Methods¶
The pressure on the pharmaceutical industry to cut drug development costs triggered the development of ligand-based drug design methods, of which the "molecular similarity principle" has proven to be useful. Computational methods based on the molecular similarity principle are generally cheaper and faster than experimental methods. In the chart below you can see the costs to get a new drug on the market over the last few years, along with the number of new drugs on the market.
articles
The price of innovation: new estimates of drug development costs J.A. DiMasi et al. J. Health Econ. 22 2003
Perspectives¶
The concept of molecular similarity has nurtured the imagination and creativity of several generations of medicinal chemists. Because of the fuzziness of the concept medicinal chemists live with the contradiction that simultaneously similar compounds do have and do not have similar activity. In this combinatorial chemistry era, it remains a cheminformatics challenge to create more refined tools enabling the medicinal chemist to navigate in this fuzziness and exploit novel values that were previously ignored.

Copyright © 2024 drugdesign.org