Ligand-Based Approaches¶
Info
There are four methods for designing new molecules in a pharmacophore-based drug design approach. Each method is presented and illustrated with concrete examples.
Number of Pages: 63 (±1 hours read)
Last Modified: January 2006
Prerequisites: Principles of Rational Drug Design, Pharmacophore-Based Drug Design: Analysis
Introduction¶
Beginning the Design Phase¶
Once the phase of analysis is complete, the design phase can proceed. New lead compounds are designed based on the pharmacophore model obtained in the previous analyses. The aim of the design is to convert all the generated knowledge into candidate molecules to be synthesized (or ordered) for biological testing.

Creativity of the Design¶
The knowledge accumulated during the phase of analysis determines the success of the design. However, creativity is extremely important as well. The creativity and imagination convert knowledge into 'intelligent' molecules and initiate scientific breakthroughs.

Good Control of the Conformational Features¶
In all design actions, it is of paramount importance not to overlook the conformational consequences of the planned action. Breaking a ring or introducing a substituent or spacer are all operations that have consequences in the conformational preferences of the designed candidate. The two following examples of butaclamol and staurosporine illustrate the consequences of a design where the conformational implications were mismanaged.

Butaclamol Example (Bad Design & Bad Results)¶
Let's suppose that someone intends to design mimics of butaclamol and decide to remove a ring by deleting one bond in the structure of this compound.
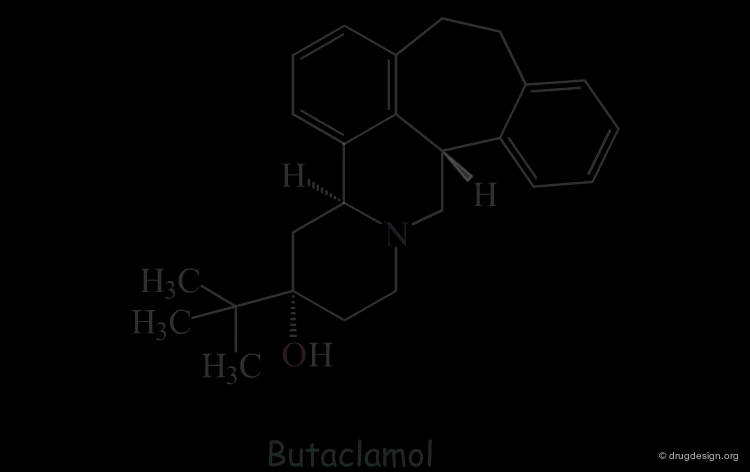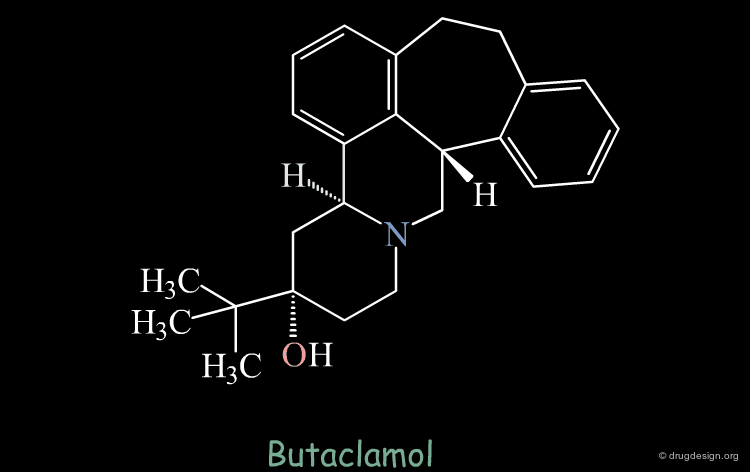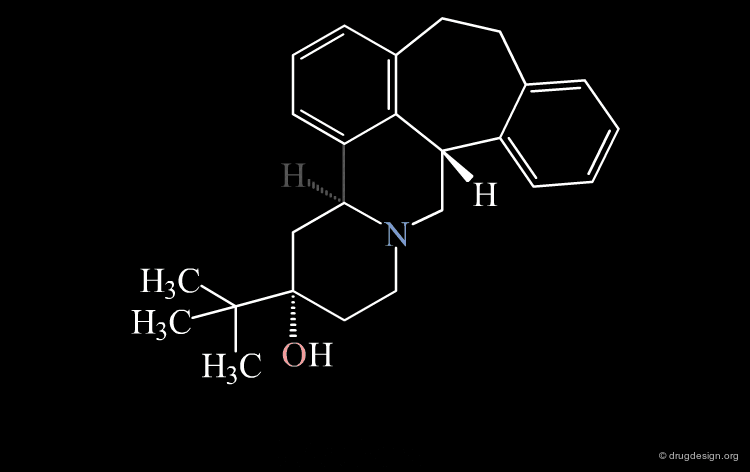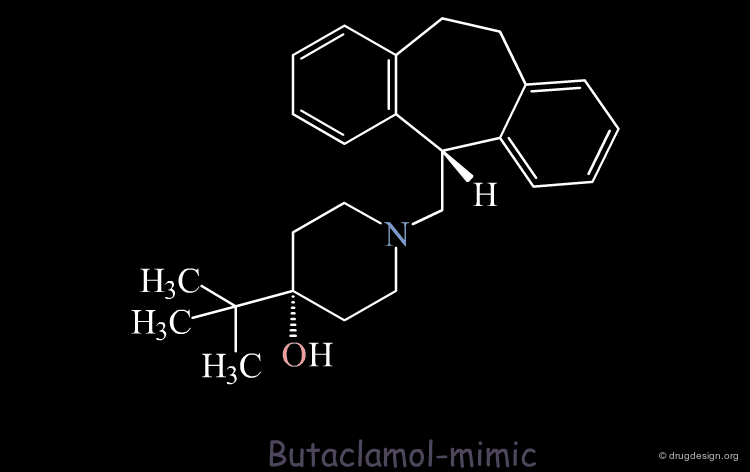
Intuitive Justification of the Design¶
The validity of this hypothesis could be justified by the good superimposition of models of the two structures. But this manipulation is just an exercise that is far from reality.

Strong van der Waals Repulsions¶
In the designed compound with the unmodified conformation (conformation A), there exist extremely repulsive van der Waals interactions between the hydrogens connected to the carbon atoms on which the bond was removed.

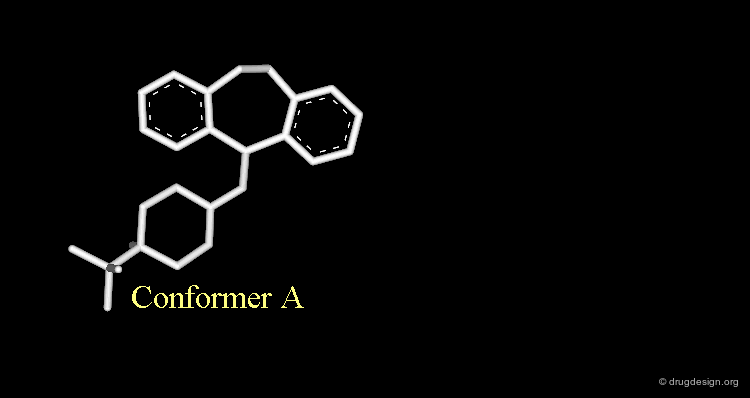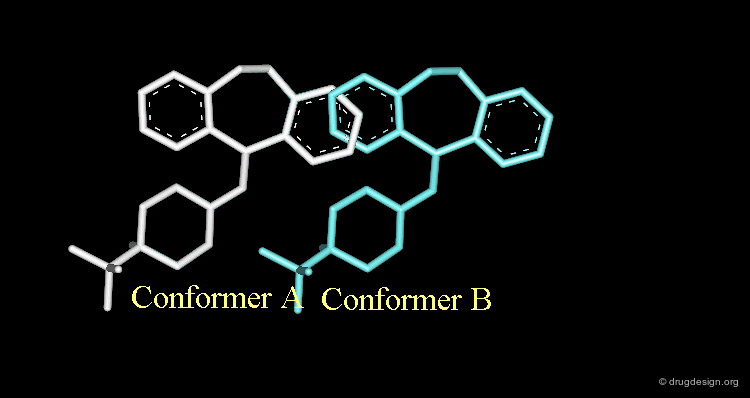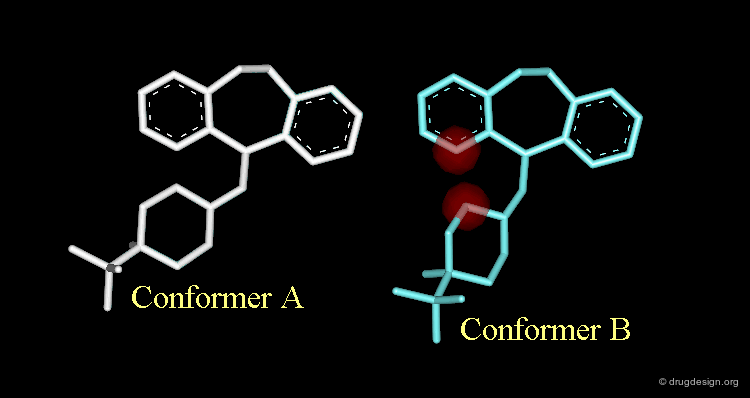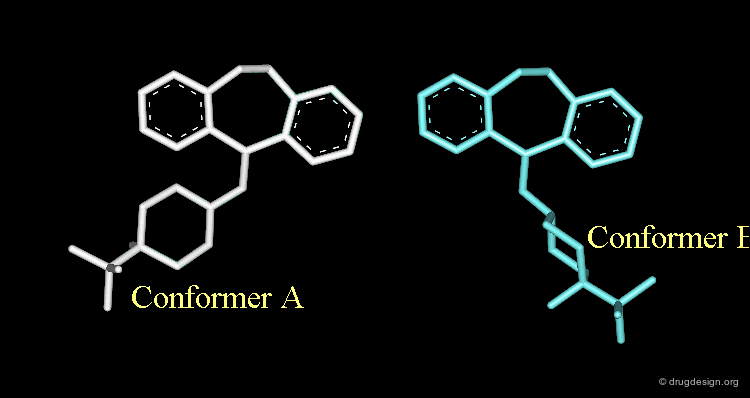
Conformational Rearrangement¶
These repulsions will induce a rotation of the tricyclic moiety relative to the piperidine ring creating a more stable but completely different conformation (conformation B).
The Designed Molecule is a Poor Butaclamol 3D Mimic¶
The designed compound is no longer superimposed on top of the structure of the butaclamol and the validity of undertaking a synthetic effort for such a poor butaclamol mimic is questionable.

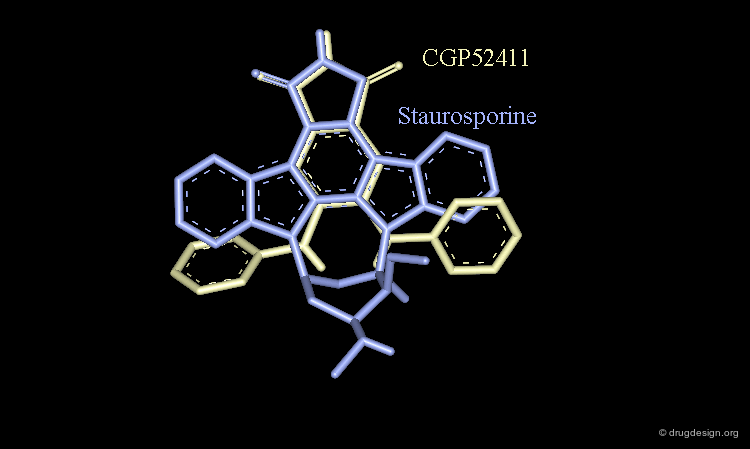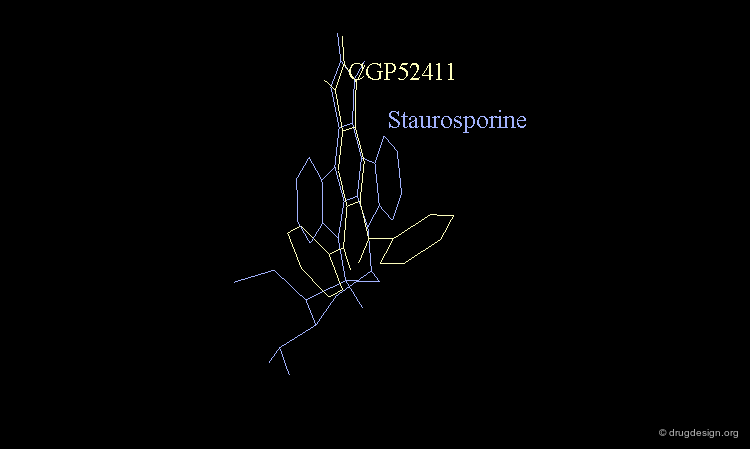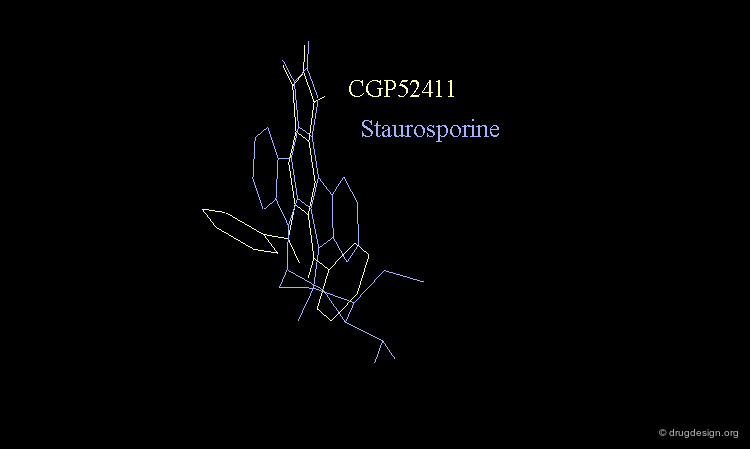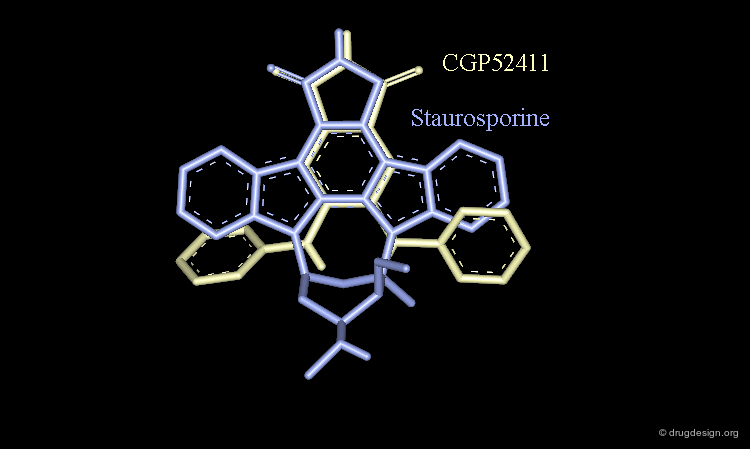
Staurosporine Example (Bad Design & Good Results)¶
Staurosporine is a competitive inhibitor of ATP against many protein kinases. Looking for new EGF-R kinase inhibitors, the structure of CGP52411 was conceived by removing rings and bonds from the 2D formula of staurosporine. The molecule proved to be a EGF-R kinase competitive inhibitor of ATP (IC50 = 0.3 µM).

articles
4,5-Dianilinophthalimide: a Protein-Tyrosine Kinase Inhibitor with Selectivity for the Epidermal Growth Factor Receptor Signal Transduction Pathway and Potentin vivo Antitumor Activity Buchdunger E, Trinks U, Mett H, Regenass U, Muller M, Meyer T, McGlynn E,Pinna LA, Traxler P, and Lydon NB Proc. Natl. Acad. Sci. USA. 91 1994
Staurosporine does not Mimic ATP¶
Staurosporine is a competitive inhibitor of ATP because it is anchored to the same residues of the catalytic site of the protein kinase enzyme with the formation of two hydrogen bonds (bidentate). However, the molecule does not mimic the structure of ATP; it only hinders the anchorage of ATP to the same residues of the catalytic site.

CGP52411 is Not a Mimic of Staurosporine¶
CGP52411 is not a mimic of staurosporine. The latter has a flat molecular geometry due to its polycyclic structure. CGP52411 is not flat and the two anilino moieties are not stable as they are in staurosporine. They must rotate until the van der Waals repulsions between the anilino and the phtalimido moieties have been relieved.

Unexpected Favorable 3D Rearrangement¶
In summary, staurosporine is neither a mimic of ATP nor a mimic of CGP52411. Therefore one cannot expect CGP52411 to be a mimic of ATP. However by chance, the conformational rearrangement of CGP52411 positioned one of the phenyl moieties that rotated exactly at the same place of the sugar fragment of ATP. Finally a mimic of ATP, CGP52411 was found. However from the design point of view, this is not the result of good design considerations but of chance.
Obvious Design¶
Sometimes the information discovered through the analysis phase is precise enough that there is no need to look for methods to decide which molecule to prepare. With some intelligence the medicinal chemist can rapidly "see" what molecule to do. Good pharmacophore models carry the necessary information to undertake such "obvious design" operations. One can design a molecule by the "hybrid" assembling elements of different molecules.

The Four Design Methods¶
The Four Design Methods¶
In pharmacophore-based drug design, there are four different ways of designing new molecules: chemical manipulation and modification, manual design, database searching, and finally computerized de novo design.

Chemical Modifications¶
Principles of Analog Design¶
In an analog design program, it is important to avoid introducing several simultaneous changes to the lead reference molecule. Experience shows that useful information can be obtained only when single structural changes in the lead molecule are involved. Changes may be bioisosteric replacements, design of rigid analogs, homologation of alkyl chains, alteration of ring size, etc...
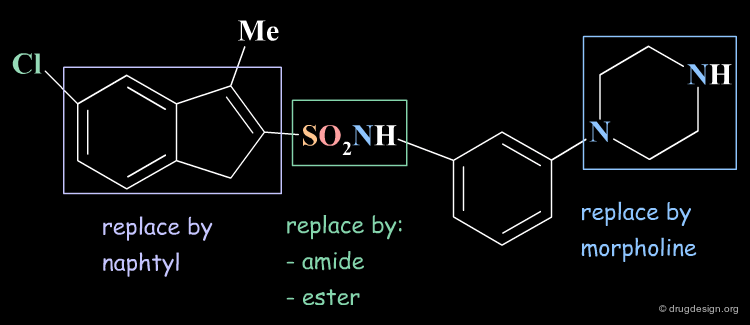
Bioisosteric Replacements: Principle¶
Bioisosterism is a concept that derives from the observation that chemically unrelated compounds may have very similar physicochemical properties. Bioisosters are chemical groups that have similar physical properties. Molecules that are designed by bioisosteric replacements are expected to have similar biological properties.




articles
Isosterism and Bioisosterism in Drug Design Burger A Prog. Drug Res. 37 1991
Isosterism and Bioanalogy in Drug Design Burger A Med. Chem. Res. 4 1994
Isosterism and Bioisosterism Case Studies with Muscarinic Agonists Floersheim P, Pombo-Villar E and Shapiro G Chimia 46 1992
Bioisosterism in Drug Design Lipinski CA Ann. Rev. Med. Chem. 21 1986
The Use of Bioisosteric Groups in Lead Optimization Olesen PH Curr. Opin. Drug. Discov. Devel. 4 2001
Bioisosterism: A Rational Approach in Drug Design Patani GA and LaVoie EJ Chem. Rev. 96 1996
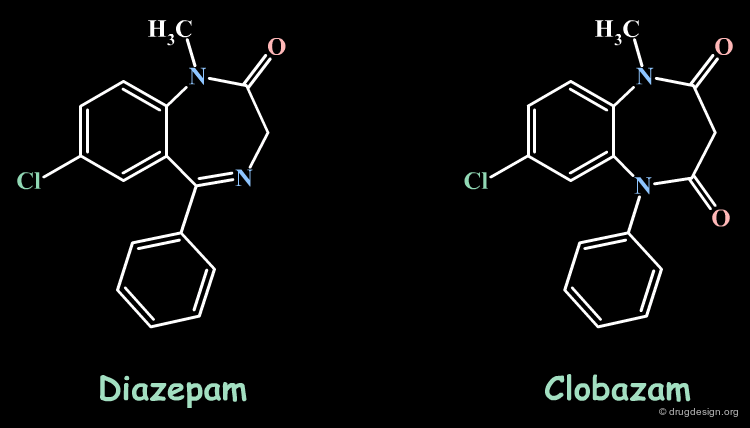
Bioisosteric Replacements: Diazepam¶
Clobazam was designed by replacing the imino group of diazepam by the isosteric amide equivalent. The two molecules have similar molecular geometries and lipophilicity. Like diazepam, clobazam is an anxiolytic drug.
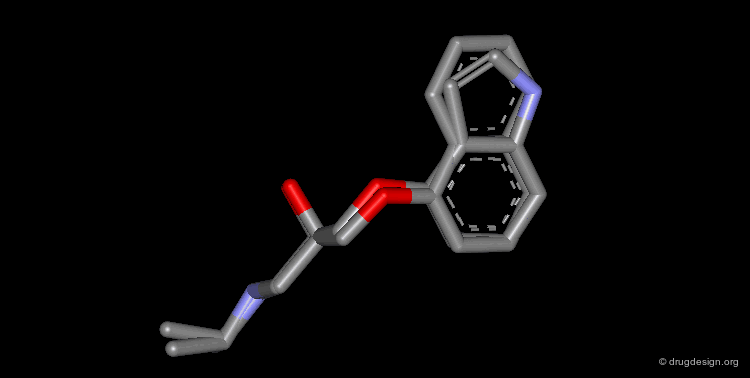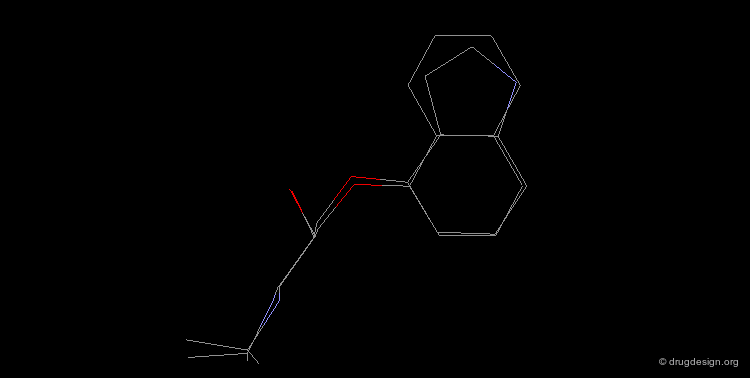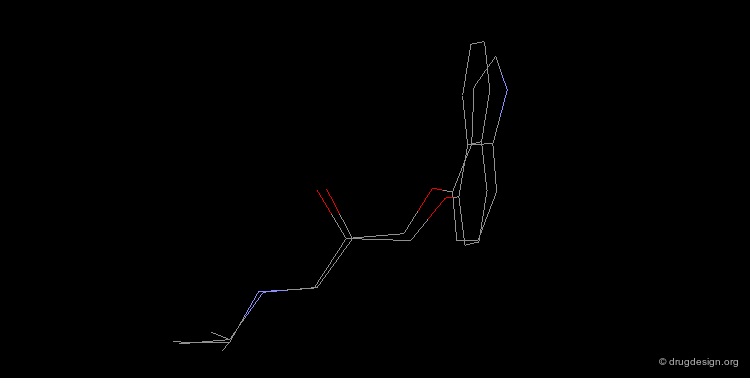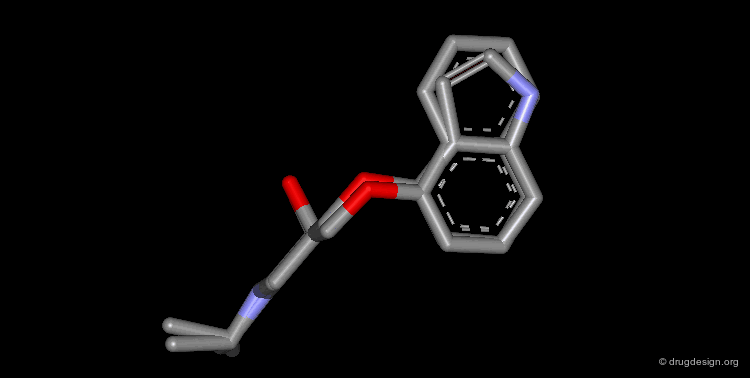
Bioisosteric Replacements : Beta-Blockers¶
The efficiency of propranolol, which is a β-blocker, was improved by replacing the naphthyl group by an indolyl group (pindolol).

Rigid Analogs: Principle¶
To increase biological activity some degree of molecular rigidity can be imposed to an active flexible molecule by incorporating its structural elements in a constrained cyclic system. The key functional groups are then constrained to a limited range of steric positions and interatomic distances.

articles
Lowering the Entropic Barrier for Binding Conformationally Flexible Inhibitors to Enzymes Khan AR, Parrish JC, Fraser ME, Smith WW, Bartlett PA, and James MN Biochemistry 37 1998
Energetic and Entropic Factors Determining Binding Affinity in Protein-Ligand Complexes Klebe G, Bohm HJ J. Recept Signal Transduct Res. 17 1997
The Cost of Conformational Order: Entropy Changes in Molecular Associations Searle MS and Williams DH J. Am. Chem.Soc. 14 1992
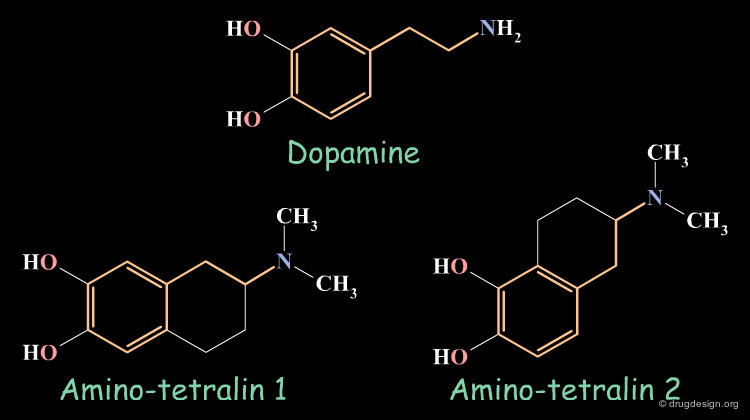
Rigid Analogs : Dopaminergics¶
Dopamine has two trans rotameric forms that can be incorporated in different structurally constrained systems. Different biological activities were observed for the following tetralin rigid analogs, they give an indication on the conformations assumed by the flexible dopamine molecule when it binds to different sub-populations of dopamine receptors.
articles
Further Characterization of Structural Requirements for Agonists at the Striatal Dopamine D-1 Receptor. Studies with a Series of Monohydroxyaminotetralins on Dopamine-Sensitive Adenylate Cyclase and a Comparison with Dopamine Receptor Binding Seiler MP and Markstein R Mol Pharmacol. 22 1982
Further Characterization of Structural Requirements for Agonists at the Striatal Dopamine D2 Receptor and a Comparison with those at the Striatal Dopamine D1 receptor. Studies with a Series of Monohydroxyaminotetralins on Acetylcholine Release from Rat Striatum Seiler MP and Markstein R Mol Pharmacol. 26 1984
book
Seiler MP, Bolsterli JJ, Floersheim P, Hagenbach A, Markstein R. Pfaffli P, Widmer A, and Wuthrich H Perspectives in Medicinal Chemistry VCH 1993
Alteration of Ring Size: Principle¶
The modification of the size of a ring can alter the molecular geometry and the relative location of the pharmacophoric elements; this can affect the biological properties of the molecule.

Alteration of Ring Size: Imipramine¶
Imipramine is a tricyclic antidepressant drug. The central seven membered ring was modified to six and to five membered ring structures. Dimetacrine has a central six-membered ring, and this drug has an antidepressant action comparable to that of imipramine.


Ring Suppression: Principle¶
The suppression of a ring in the formula of an active compound allows one to obtain related molecules with different structures. However one has to be aware that the molecular geometries of the reference and the designed molecules might not be superimposable in 3D.

Ring Suppression: Doxepin¶
The structure of doxepin has been used as a starting point for the design of non-polycyclic analogs. By cuting one of the bonds in this structure results in a doxepin-like analog that has one ring less.

Homologation of Alkyl Chains: Principle¶
The progressive increase in the number of carbon atoms in a chain progressively increases the lipophilic character of the molecule and changes the partition coefficient. This can affect the absorption, distribution, metabolism or excretion properties of the molecule.
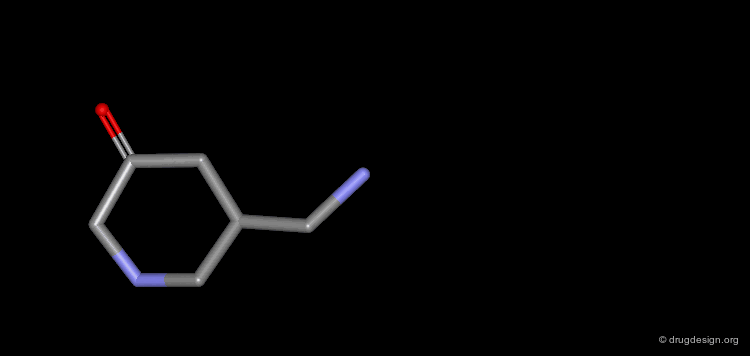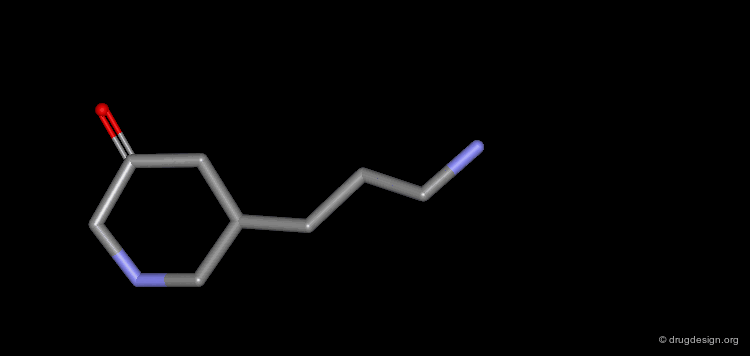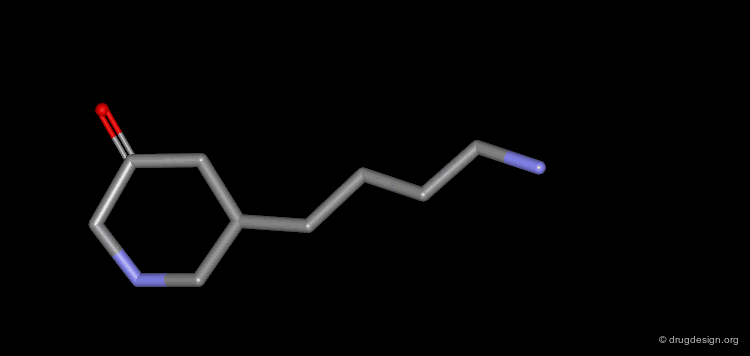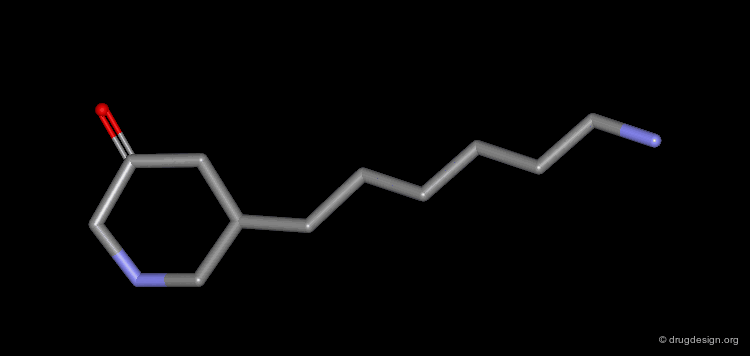
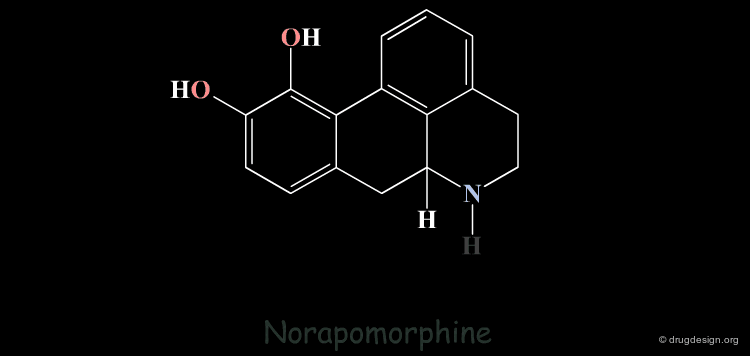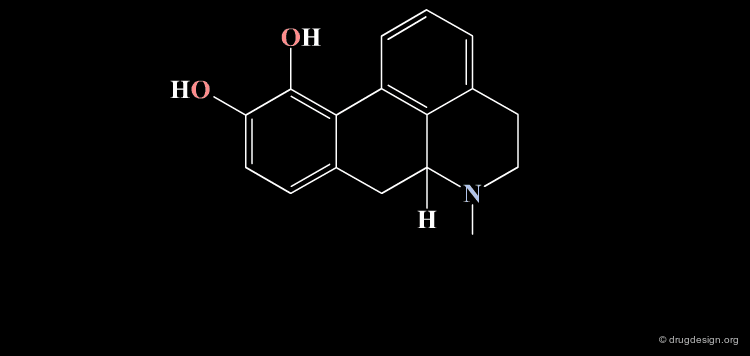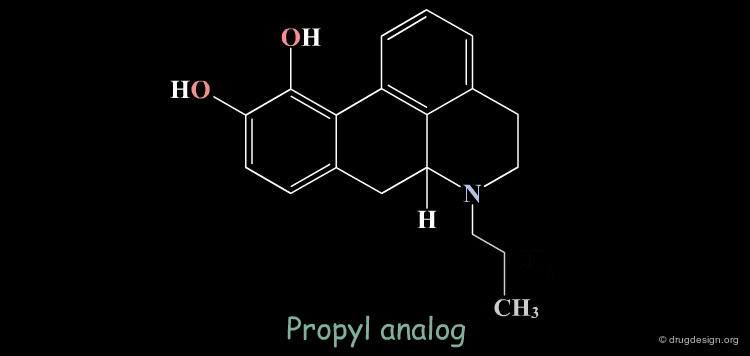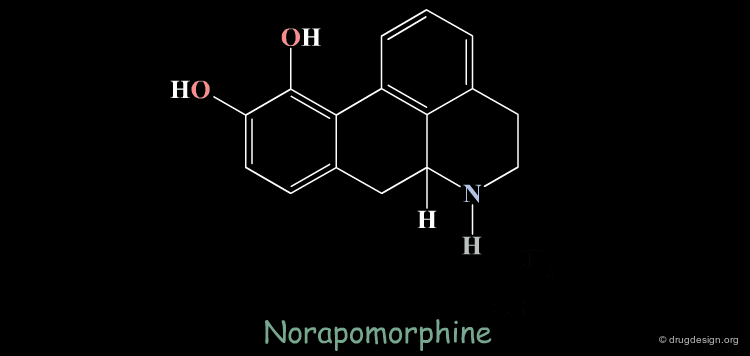
Homologation of Alkyl Chains: Apomorphine¶
The hydrogen atom connected to the nitrogen of apomorphine was replaced by methyl, ethyl, n-propyl and n-butyl groups. The best activity was found for the n-propyl analog, whereas the n-butyl demonstrated a great loss in potency.
Alteration of Stereochemistry: Principle¶
Alterations of stereochemical centers in an active molecule may lead to similar or to completely different biological activities.

Alteration of Stereochemistry: Progesterone¶
The naturally occurring steroid hormones consist of a fused ring system with stereochemistries as indicated in the structure of progesterone. Synthetic analogs with modified stereochemistries at different chiral centers were prepared but did not lead to therapeutically interesting molecules, except for a special class of steroids called retrosteroids. Retroprogesterone is an example of a synthetic analog that is more active than the natural progesterone.

articles
Enantiomeric Steroids: Synthesis, Physical and Biological Properties Biellmann JF Chem. Rev 103 2003
Title Westerhof P Rec. Trav. Chim 79 1960
A new class of hormonally active steroids Rwwrnik EH, Scholer HF, Westerhof P, Querido A, Kassenaar AA, Diczfalusy E, Tillinger KC Naturte 186 1960
The x-ray structure analysis of 17-beta-bromoacetoxy-9-beta, 10-alpha-androst-4-en-3-on Oberhansli WE, Robertson JM Helv. Chim. Acta 50 1967
book
Miyake T and Rooks WH II Methods in Hormone Research Vol V, Part C Academic Press NY 1966
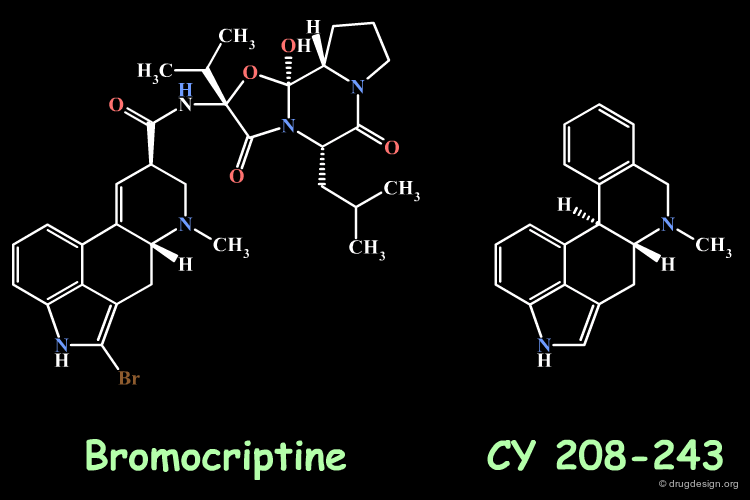
Homologation by Simplification: Bromocryptine¶
Ergot alkaloids like bromocryptine have been the starting point for preparing simplified synthetic analogs. CY 208-243 is an example of a simplified bromocryptin and is a dopamine receptor agonist. The simplification of a reference molecule is a general method in analog design and may lead to molecules that are more potent than the initial lead structure.
Altering Interatomic Distances¶
The systematic alteration of interatomic distances between pharmacophore elements allows us to both explore the requirements of a receptor site and to optimize a series of molecules. The following is a typical analog program conducted for the systematic exploration of the biological properties of a reference active compound.

Aromatic Ring Position Isomers: Principle¶
It has been observed that small structural changes as the modification of the substitution of an aromatic ring can lead to important changes in the biological activities of a series. The substituents of a reference molecule with an aromatic system can be modified systematically.

Aromatic Ring Position Isomers : β-Adrenergic Drugs¶
β-adrenergic agents having a phenethanolamine moiety are chemically related to the natural hormones adrenaline and nor-adrenaline. It has been observed that small structural changes such as the modification of the substitution of the aromatic ring can transform an antagonist into an agonist and vice versa. The following example shows that moving the sulfonamide group from meta to para changed an agonist into an antagonist.

Chemical Modifications for SAR Information¶
The systematic exploration of analogs of a given structure allows one to obtain information and accumulate data that are of great utility for understanding the structure-activity relationships.

articles
Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins Cramer RD, Patterson DE and Bunce JD J Am. Chem. Soc. 110 1988
A Mathematical Contribution to Structure Activity Studies Free SM and Wilson J J. Med. Chem. 7 1964
A Quantitative Approach to Biochemical Structure-Activity Relationships Hansch, C Acc. Chem. Res. 2 1969
Molecular Graphics and QSAR in the Study of Enzyme-Ligand Interactions Hansch C and Klein T Acc. Chem. Res. 19 1986
Quantitative Structure-Activity Relationships and the Unnamed Science Hansch C Acc. Chem. Res. 26 1993
Quantitative Structure-Activity Relationships. 1. The Modified Free-Wilson Approach Kubinyi H and Kehrhahn OH J. Med. Chem. 19 1976
QSAR and 3D QSAR in Drug Design Part 1: Methodology Kubinyi H Drug Discovery Today 2 1997
Database Searching¶
3D Database Searching¶
3D database searching enables one to identify existing molecules that match a pharmacophore hypothesis. The 3D structure of small molecules can be obtained from various sources like crystallographic databases (e.g. CSD), commercially available databases (e.g. FCD) or computerized model-builders (e.g. CONCORD). It is also possible to identify molecules that possess similar 3D shapes. An advantage of this method is that it generally does not require special synthetic efforts (the molecule is known) but on the other hand, it is highly dependent on the content of the database.

articles
The Cambridge Structural Database: a Quarter of a Million Crystal Structures and Rising Allen FH Acta Cryst. B58 2002
Selected Concepts and Investigations in Compound Classification, Molecular Descriptor Analysis, and Virtual Screening Bajorath J J. Chem. Inf. Comput. Sci. 41 2001
Similarity Searching in Databases of Chemical Structures Downs GM and Willet P Rev. Comput. Chem. 7 1995
Using CONCORD to Construct a Large Database of Three-Dimensional Coordinates from Connection Tables Rusinko A III, Sheridan RP, Nilakantan R, Haraki KS, Bauman N and Venkataraghavan R J. Chem. Inf. Comput. Sci. 29 1989
3DSEARCH: A System for Three-Dimensional Substructure Searching Sheridan RP, Nilakantan R, Rusinko A III, Bauman N, Haraki KS and Venkataraghavan R J. Chem. Inf. Comput. Sci. 29 1989
Searching for Pharmacophores in Large Coordinate Data Bases and its Use in Drug Design Sheridan RP, Rusinko A III, Nilakantan R and Venkataraghavan R Proc. Natl. Acad. Sci. USA. 86 989
Chemical Similarity Searching Willett P, Barnard JM, and Downs GM J. Chem. Inf. Comput. Sci. 38 1998
Problems of Conformational Complexity¶
Molecules are flexible entities. The bioactive conformation of a given compound is one form among the many possible forms of the molecule. Even with the remarkable progress in computer storage technology, it is absolutely impossible to imagine that one could store in a databank all the thousands of possible conformers of every single molecule present in the database. In more recent systems conformational flexible searching has started to be considered. In this case the databank contains one or two conformations for each molecule, however a large number of flexible forms are generated "on the fly". In this approach the limitation is not anymore on the storage capacity but on the computational capabilities.

articles
Rapid Calculation of Coordinates from Distance Matrices Crippen GM J. Comput. Phys. 26 1978
Use of Flexible Queries for Searching Conformationally Flexible Molecules in Databases of Three-Dimensional Structures Guner OF, Henry DR and Pearlman RS J. Chem. Inf. Comput. Sci. 32 1992
Three-Dimensional Shape-Based Searching of Conformationally Flexible Compounds. Hahn MA J. Chem. Inf. Comput. Sci. 37 1997
Flexible 3D Searching: The Directed Tweak Technique Hurst T J. Chem. Inf. Comput. Sci. 34 1994
Distance Geometry Kuntz ID, Thomason JF, Oshiro CM Methods Enzymol. 177 1989
Pharmacophoric Pattern Matching in Files of Three-Dimensional Chemical Structures: Comparison of Conformational-Searching Algorithms for Flexible Searching Clark DE, Jones G, Willett P, Kenny PW, and Glen RC J. Chem. Inf. Comput. Sci. 34 1994
book
Crippen GM Distance Geometry and Conformational Calculations Research Studies Press, Wiley 1981
Crippen GM and Havel TF Distance Geometry and Molecular Conformation Wiley 1988
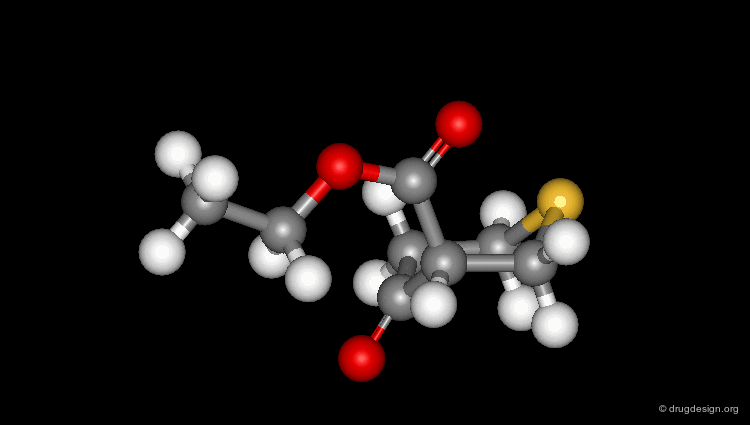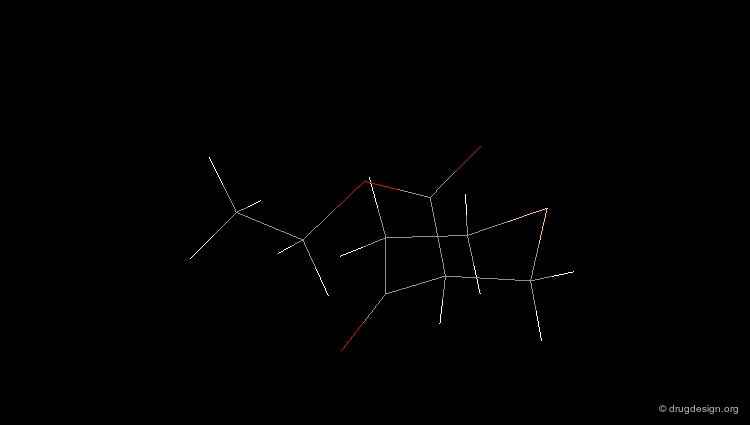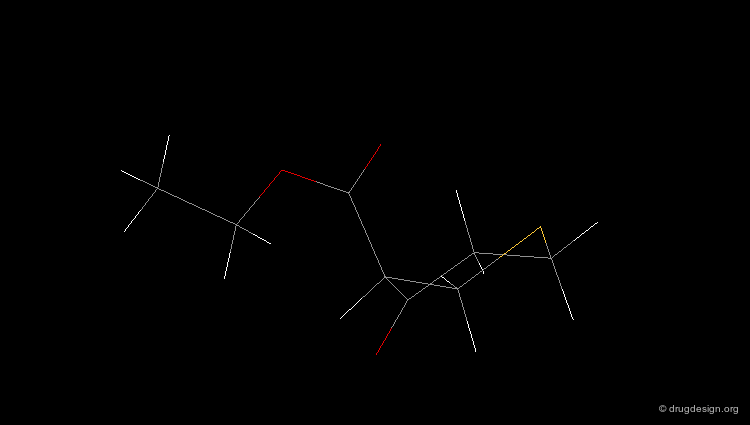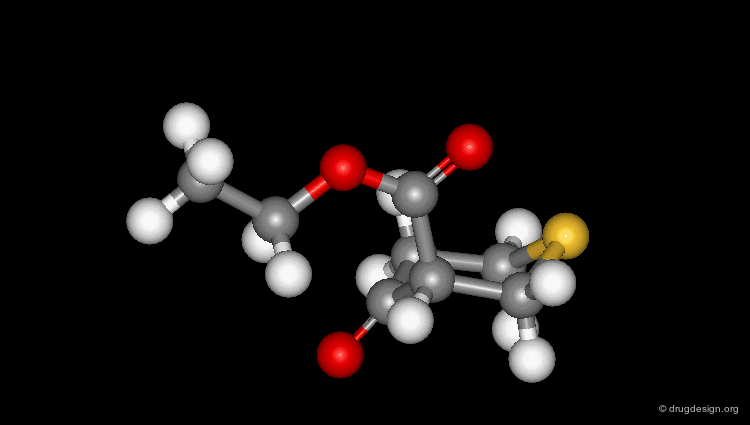
Check Molecular Geometry of a Hit¶
Whenever a hit is obtained in a database search, one should always be careful and check its geometry. Databases constructed with 2D->3D converters may contain molecules with poor quality geometries. Let's look at the 'hit' found in a database search in the following example. Not only is the substituent in an axial orientation, but the geometry of the ester fragment is "s-cis", a form that is extremely improbable in acyclic ester moieties.
Example of 3D Searching: Pharmacophore Query¶
The involvement of protein kinase C (PKC) enzymes in uncontrolled cellular proliferation, makes them attractive targets for chemotherapeutic intervention against cancer. A computerized 3D-database search on more than 200,000 structures was performed using the following pharmacophore which was deduced from a series of known active phorbol esters. PDBU is an example of a member of this series.

articles
The Discovery of Novel, Structurally Diverse Protein Kinase C Agonists through Computer 3D-Database Pharmacophore Search. Molecular Modeling Studies Wang S, Zaharevitz DW, Sharma R, Marquez VE, Lewin NE, Du L, Blumberg PM, and Milne GW J. Med. Chem. 37 1994
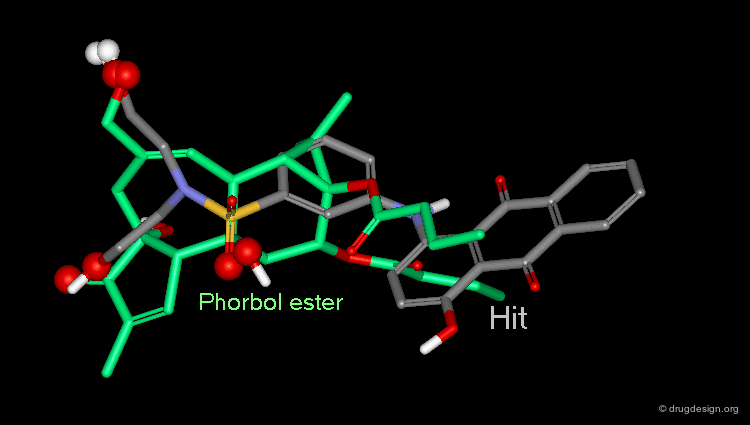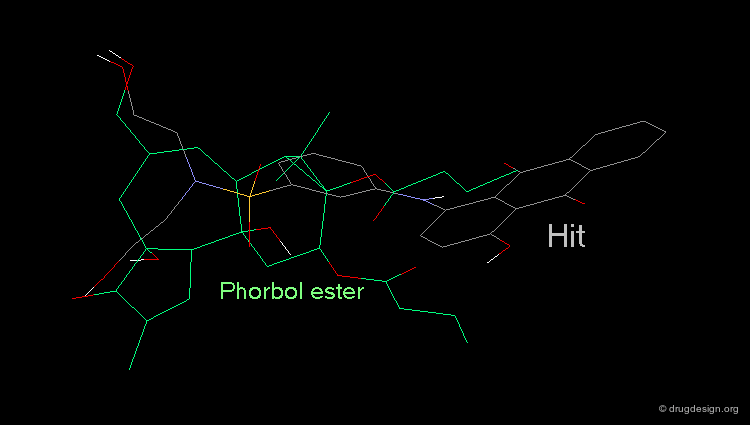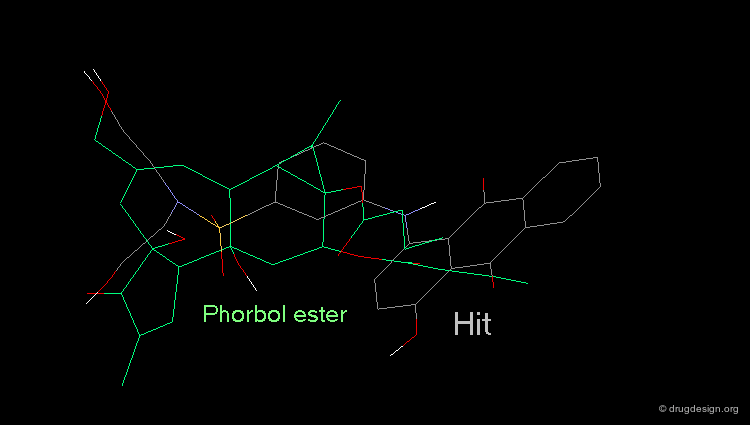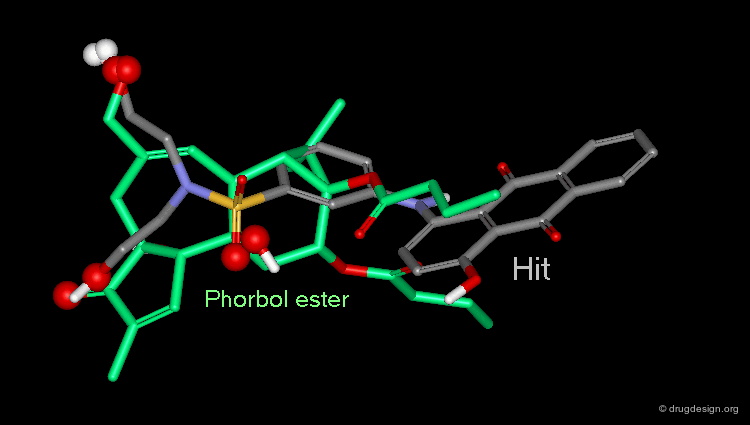
Hit Obtained by 3D Database Searching¶
This 3D database search led to the discovery of new potent PKC agonists chemically unrelated to the reference phorbol esters such as the quinone derivative.

articles
The Discovery of Novel, Structurally Diverse Protein Kinase C Agonists through Computer 3D-Database Pharmacophore Search. Molecular Modeling Studies Wang S, Zaharevitz DW, Sharma R, Marquez VE, Lewin NE, Du L, Blumberg PM, and Milne GW J. Med. Chem. 37 1994
Example of 3D Database Searching: Shape Query¶
Since shape complementarity is a very important consideration in drug-receptor interactions, it is useful to search a 3D database for similar 3D shapes with a reference molecule. If we search for compounds having a similar shape to PDBU we get several hits in which a similarity in the 3D shape is found, within a similarity threshold factor which can be fixed. The following is an example of a database hit visualizing the similarity between the 3D shape from PDBU and the found molecule.

articles
Similarity Searching In Files of Three-Dimensional Chemical Structures: Comparison of Fragment-Based Measures of Shape Similarity Bath PA, Poirrette AR, Willett P and Allen FH J. Chem. Inf. Comp. Sci. 34 1994
New Molecular Shape Descriptors: Application In Database Screening Good AC, Ewing TJA, Gschwend DA and Kuntz ID J. Comput. Aided Mol. Des. 9 1995
Three-Dimensional Shape-Based Searching of Conformationally Flexible Compounds Hahn M J. Chem. Inf. Comput. Sci. 37 1997
Similarity of Molecular Shape Meyer AY and Richards WG J. Comput. Aid. Mol. Des. 5 1991
book
Mezey PG Molecular Similarity in Drug Design Blackie 1995
Molecules Obtained by Shape Searching¶
Although the similarity of the 3D shape of PDBU and the database hit (a quinoline derivative) is high, there is a vast difference in their chemical structure. This type of database search complements the powerful pharmacophore-based database search. Searching for shape similarity is useful only if the pharmacophore is properly incorporated.

articles
Similarity Searching In Files Of Three-Dimensional Chemical Structures: Comparison Of Fragment-Based Measures Of Shape Similarity Bath PA, Poirrette AR, Willett P and Allen FH J. Chem. Inf. Comp. Sci. 34 1994
New Molecular Shape Descriptors: Application In Database Screening Good AC, Ewing TJA, Gschwend DA and Kuntz ID J. Comput. Aided Mol. Des. 9 1995
Three-Dimensional Shape-Based Searching of Conformationally Flexible Compounds Hahn M J. Chem. Inf. Comput. Sci. 37 1997
Similarity of Molecular Shape Meyer AY and Richards WG J. Comput. Aid. Mol. Des. 5 1991
book
Mezey PG Molecular Similarity in Drug Design Blackie 1995
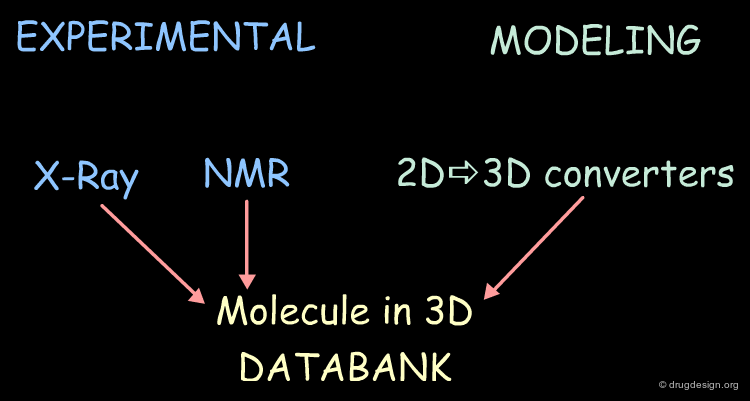
Databases of Molecules in 3D¶
The 3D structures of molecules can be either determined experimentally (X-ray data, NMR) or by modeling techniques (2D->3D converters).
book
Priestle JP and Paris CG Guidebook in Molecular Modeling and Drug Design Academic Press 1996
Databases of Commercial Molecules¶
Throughout the world, there are thousands of chemical suppliers offering more than 3 million organic compounds. Soon combinatorial chemistry will lead to an explosion of this number. The advantage of using commercially available molecules in database searching is that they can be purchased and tested rapidly. This accelerates the lead discovery process. In order to conduct the searching in 3D, model-builders are used to convert the 2D catalogs into 3D databases.

De-Novo Design¶
Automated Construction Approaches¶
Designing molecules that assemble a set of disconnected functional groups in 3D is not a trivial task. The purpose of de novo automated construction approaches is to find appropriate spacers to assemble these disconnected elements. These spacers are made of small units like atoms, functional groups or fragments. The solutions show alternatives for positioning the same key fragments and therefore provide structural diversity.

articles
SPROUT: 3D Structure Generation Using Templates Mata P, Gillet VJ, Johnson AP, Lampreia J, Myatt GJ, Sike S, and Stebbings L J. Chem. Inf. Comput. Sci. 35 1995
CAVEAT: a Program to Facilitate the Design of Organic Molecules Lauri G and Bartlett PA J Comput. Aided Mol Des. 8 1994
The NEWLEAD Program: A New Method for the Design of Candidate Structures from Pharmacophoric Hypotheses Tschinke V and Cohen NC J. Med. Chem. 36 1993
PRO_LIGAND: An Approach to de Novo Molecular Design. 2. Design of Novel Molecules from Molecular Field Analysis (MFA) Models and Pharmacophores Waszkowycz B, Clark DE, Frenkel D, Li J, Murray CW, Robson B and Westhead DR J. Med. Chem. 37 1994
Algorithm Based Approaches¶
Assembling together pharmacophoric elements can be done efficiently with computerized construction programs. In some systems, the new molecule is constructed atom-by-atom while other systems incorporate a library of pre-prepared fragments ready to be assembled. Whatever algorithms are used, the input of such programs is the pharmacophore considered as the '3D query', and the output consists of possible solutions, namely different molecules carrying the query pharmacophore in the proper geometry.

articles
SPROUT: 3D Structure Generation Using Templates Mata P, Gillet VJ, Johnson AP, Lampreia J, Myatt GJ, Sike S, and Stebbings L J. Chem. Inf. Comput. Sci. 35 1995
CAVEAT: a Program to Facilitate the Design of Organic Molecules Lauri G and Bartlett PA J Comput. Aided Mol Des. 8 1994
The NEWLEAD Program : A New Method for the Design of Candidate Structures from Pharmacophoric Hypotheses Tschinke V and Cohen NC J. Med. Chem. 36 1993
Example of Construction Approach¶
5-α-reductase inhibitors, which inhibit the metabolism of testosterone into dihydrotestosterone, are potent agents against prostate hyperplasia. However, most of these agents are of a steroidal type. In order to avoid all the endocrinic side effects, one can try to design non-steroid mimics.
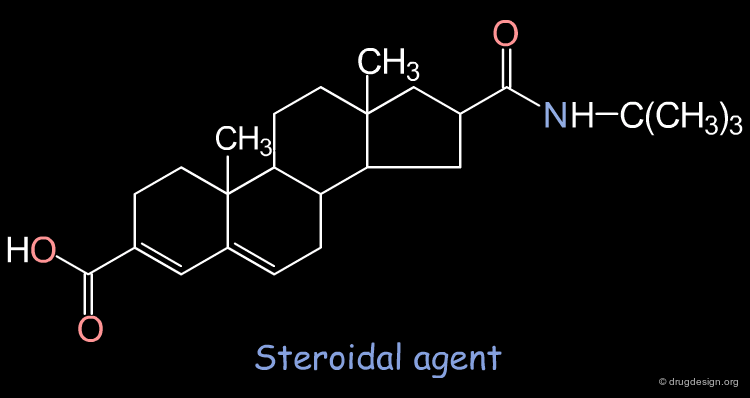
Query Pharmacophore: 5-Alfa Reductase Example¶
The construction approach NEWLEAD was used to find non-steroid analogs by linking the recognition fragments of the pharmacophore:

![]()
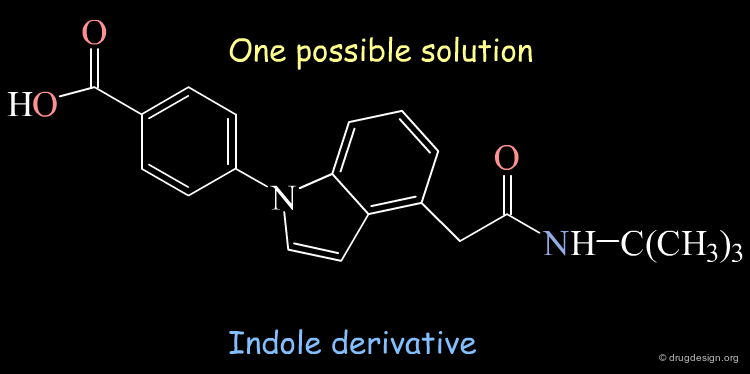
A Generated Solution¶
One of the possible solutions to the query represented is the following indole derivative: The superimposition shows how the 3D structure of the constructed indole mimics well the steroidal agent.

Automated Construction Approach: Example¶
The results of a de novo design is illustrated in the following browser. The molecules were generated by construction algorithms based on the staurosporine-like pharmacophore model as shown in the view.

Manual Design¶
Introduction to Manual Design¶
Manual design is based on deciding which molecule to consider next. Without computers, the medicinal chemist currently works like that. Trying to be consistent with the concepts of molecular design, his goal is to fully utilize the information obtained by molecules previously tested and integrate it in the structure of a new molecule to be synthesized.

Importance of the Visualization¶
In manual design the visualization is of central importance. It is important to see in a condensed way the molecular objects. This stimulates the creativity for considering other solutions and checking if the solutions designed fit with the specified constraints.

Tools in Manual Design¶
Real time rotations, molecular editing functions such as deletion, addition, fusion, connections, torsional angle alterations, superimpositions, coloring and stereo constitute the minimum necessary for sketching and assessing the validity of a solution considered in manual design. At another level energy calculations and minimizations can complete the panoply of the medicinal chemist.

Fully Exploiting the Fruits of the Analyses¶
The goal of the medicinal chemist is to fully utilize the information revealed by previously tested molecules and integrate it in the structure of a new molecule to be synthesized.

Creativity¶
This example illustrates the intelligent design of a new molecule connecting the two hydrophobic groups (phenyl and cyclohexyl) of a peptide-based molecule. The resulting molecule was biologically active. Only creativity and imagination can lead to such design.

Design of a Spacer: a Step-by-Step Process¶
The three structural moieties displayed here: phenyl, carboxyl and amino represent a pharmacophore. Assembling them in 3D into a single molecule is not obvious.

Connecting two Fragments in 3D¶
With models or modeling tools, patience, intelligence and imagination...
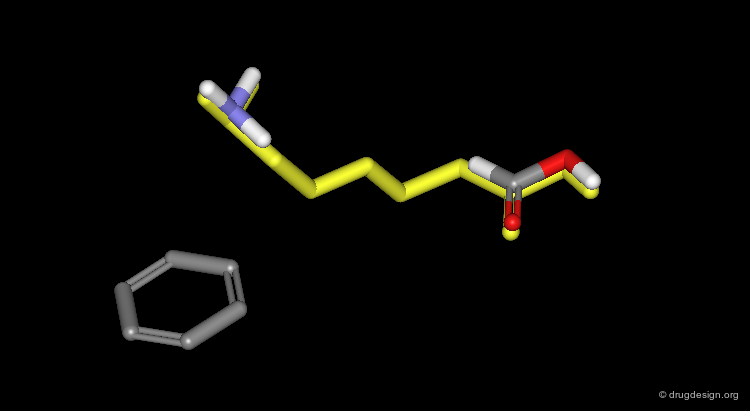
Assembly of all Fragments in 3D¶
... one can envision a solution like the one displayed here.
![]()
Manual Mimicking¶
The following example illustrates how to create an entirely new molecule using interactive manual design that imitates both the shape and the 3D distribution of the physico-chemical properties of an active reference compound.
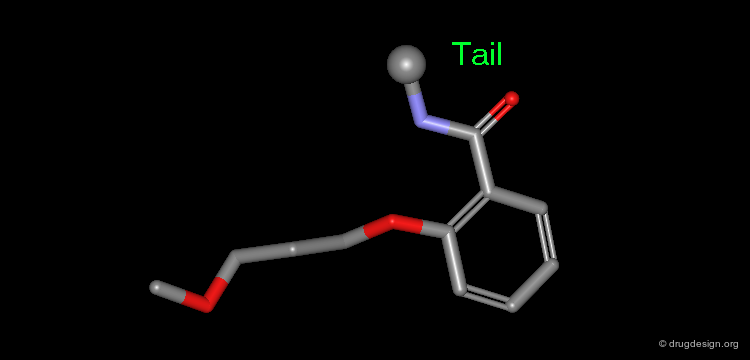
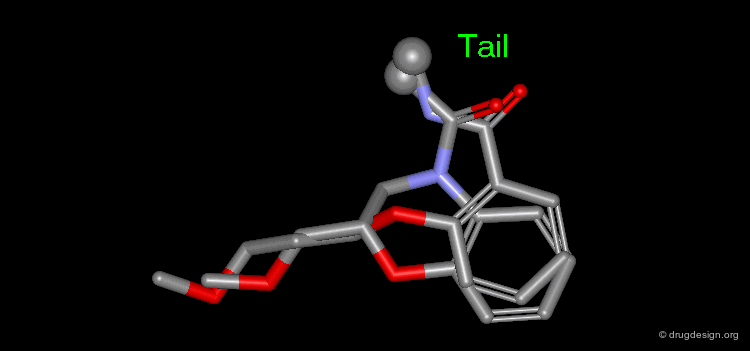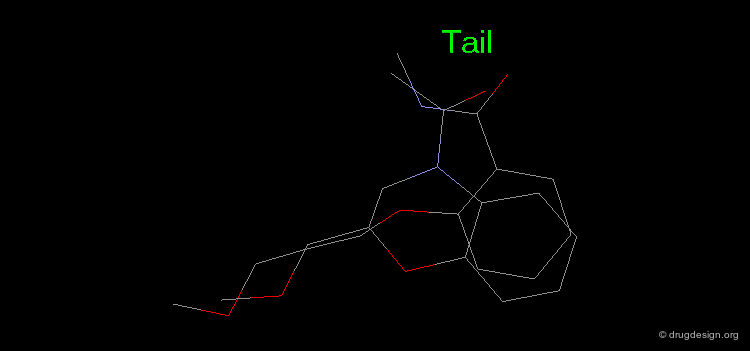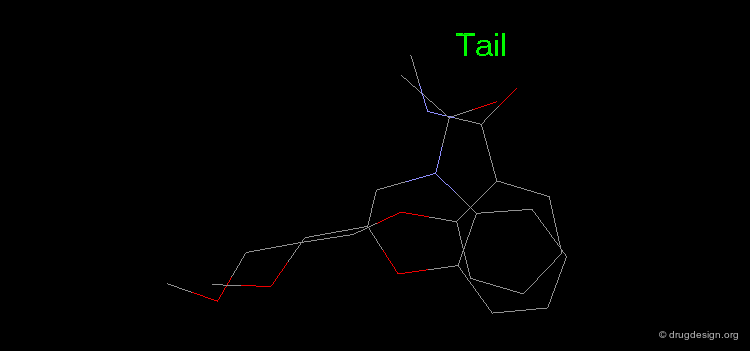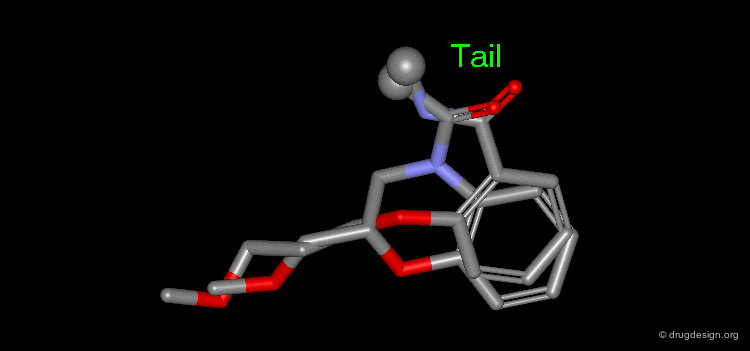
Examples of Design¶
EGF-R Protein Tyrosine Kinase Inhibitors¶
The family of the epidermal growth factor receptor (EGF-R) belongs to the larger class of the trans-membrane growth factor receptor protein kinases. Inhibitors of this enzyme therefore have great therapeutic potential in the treatment of malignant and nonmalignant epithelial diseases. Staurosporine, a microbial alkaloid was found to have some inhibitory effect competing with ATP, the cofactor of the protein kinases.
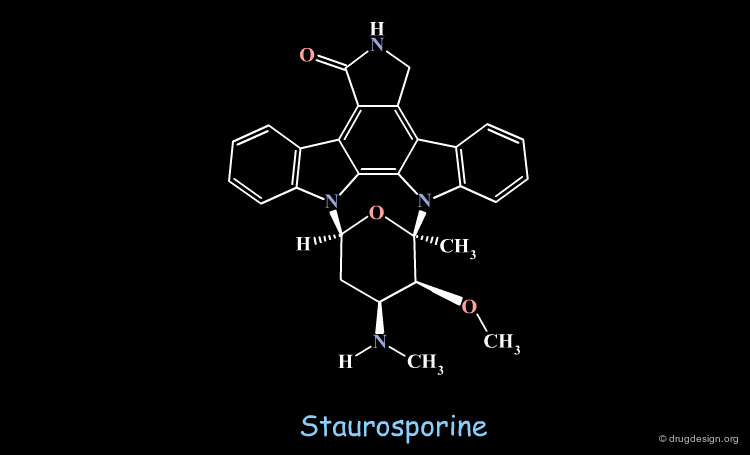
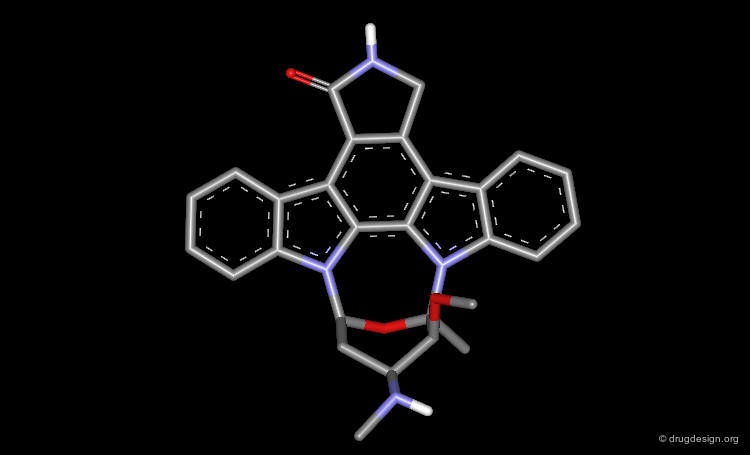
Comparing the Structures of Staurosporine and ATP¶
The analyses indicated here where made at a time when only ATP was known to bind in a bidentate manner with two hydrogen bonds and no X-ray data was available for staurosporine. ATP and staurosporine were compared in 3D despite their seemingly unrelated chemical structures. An alignment of the adenine moiety of ATP with the keto-pyrroline ring of staurosporine indicated that staurosporine exploits the same key hydrogen bond interactions as ATP.

CGP52411 a Simplified Staurosporine Molecule¶
CGP52411 was created by removing rings and bonds from the 2D formula of Staurosporine. The molecule proved to be a competitive inhibitor of ATP (IC50 = 0.3 µM) for the EGF-R protein kinase.


articles
4,5-Dianilinophthalimide: a Protein-Tyrosine Kinase Iinhibitor with Selectivity for the Epidermal Growth Factor Receptor Signal Transduction Pathway and Potent in vivo Antitumor Activity Buchdunger E, Trinks U, Mett H, Regenass U, Muller M, Meyer T, McGlynn E, Pinna LA, Traxler P, and Lydon NB Proc. Natl. Acad. Sci. USA. 91 1994
Dianilinophthalimides: Potent and Selective, ATP-Competitive Inhibitors of the EGF-Receptor Protein Tyrosine Kinase Trinks U, Buchdunger E, Furet P, Kump W, Mett H, Meyer T, Muller M, Regenass U, Rihs G, Lydon N, and Traxler P J. Med. Chem. 37 1994
Bidentate Anchorage of CGP52411¶
The alkylation of the free nitrogen of CGP52411 completely suppressed activity. This observation indicated that this compound adopts the same binding mode as staurosporine with two key bidentate hydrogen bonds. The following view illustrates an overlay of CGP52411, staurosporine and ATP; in particular it reveals a phenyl moiety of CGP52411 located in the area occupied by the ribose ring of ATP.

articles
Modelling Study of Protein Kinase Inhibitors: Binding Mode of Staurosporine and Origin of the Selectivity of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U, Traxler P J. Comput. Aided Mol. Des. 9 1995
The Design of a New EGF-R PTK Inhibitor¶
On the basis of the proposed pharmacophore model, a new series of selective EGF-R protein kinase inhibitors were designed: the pyrrolopyrimidine derivatives. 4-(m-Bromophenylamino) pyrrolopyrimidine (IC50 = 0.025 µM) is one of the most potent molecules in this series.


articles
Modelling Study of Protein Kinase Inhibitors: Binding Mode of Staurosporine and Origin of the Selectivity of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U, Traxler P J. Comput. Aided Mol. Des. 9 1995
4-(Phenylamino)Pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T, and Lydon N J. Med. Chem. 39 1996
Browser of EGF-R Protein Kinase Inhibitors¶

articles
Modelling Study of Protein Kinase Inhibitors: Binding Mode of Staurosporine and Origin of the Selectivity of CGP 52411 Furet P, Caravatti G, Lydon N, Priestle JP, Sowadski JM, Trinks U, Traxler P J. Comput. Aided Mol. Des. 9 1995
4-(Phenylamino)Pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T, and Lydon N J. Med. Chem. 39 1996
Copyright © 2024 drugdesign.org