Case Studies in 3D Mimic Design¶
Info
In what follows we present selected examples of designs based on mimicry in 3D.
Number of Pages: 65 (±1 hours read)
Last Modified: December 2008
Prerequisites:
Case Study-1 : Cimetidine Mimicry¶
Two Very Different H2-Antagonists¶
The two compounds represented here are H2-antagonists. It is difficult to imagine how they can mimic each other. On the following page we present the 3D mimicry between these two structures, a model that was exploited for the design of entirely novel antagonists.

book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
Cimetidine has a Folded Conformation¶
As revealed by X-ray analyses, cimetidine exists in a folded conformation, favored by the formation of an intra-molecular hydrogen bond between a nitrogen atom of the imidazole and a N-H group of the side chain.

book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
3D Mimicry between Cimetidine and Triazole¶
The observed biological activities can now be understood by examining this conformation for cimetidine that now mimics the triazole molecule well. Such analyses were virtually impossible by simple observation of the 2D chemical formulas of the molecules but the resulting 3D model (see button "3D mimicry") provides a good basis for the design of novel H2-antagonists.


book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
Case Study-2 : Substance P Antagonists¶
Substance P : a Ligand of CNS Receptors¶
Substance P is a member of the tachykinin family of peptides and plays a role in the transmission of pain in the central nervous system. Three different receptors for the mammalian tachykinins have been identified (NK-1, NK-2 and NK-3) and belong to the G-protein coupled receptors family. Substance P is recognized by the NK-1 receptor. Although its 3D structure is unknown, the conformation of substance P when bound to this receptor has been postulated. Here we present how this conformation has been exploited for the design of non-peptide mimics with nano-molar activities.

Conformation of Substance P¶
A model of the conformation of substance P was postulated by Convert et al., based on NMR and SAR analyses. The Novartis team recognized that mimicking the Phe7-Phe8 template (in red in the figure) was a good strategy to find non-peptide substance P antagonists.

articles
Influence of the replacement of amino acid by its D-enantiomer in the sequence of substance P. 2. Conformational analysis by NMR and energy calculations. Convert O, Duplaa H, Lavielle S, Chassaing G. Neuropeptides Aug;19(4) 1991
book
Schilling W, Bittiger H, Brugger F, Criscione L, Hauser K, Ofner S, Olpe HR, Vassout A and Veenstra S Perspectives in Medicinal Chemistry VCH 1993
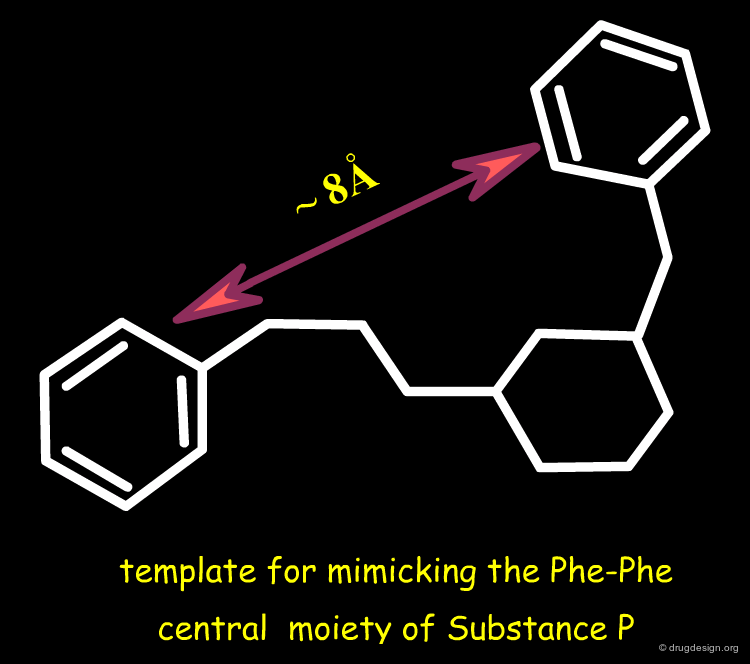
Template for Mimicking the Phe-Phe Moiety¶
A six membered ring scaffold with two 1,3 substituents was imagined as a candidate template for mimicking the 8 Å distance to unravel the phenyl rings of Phe-7 and Phe-8.
The Successful Discovery of SP Antagonists¶
Novel molecules were designed along this line and very potent Substance P antagonists were discovered. CGP47899 is an example of a successfully designed antagonist that mimics the Phe-Phe portion of the substance P substrate.


A Phe-Phe Mimic of Substance P¶
By looking at the 2D formulas of CGP47899 and Substance P, the 3D mimicry between these two structures is not immediately apparent. The central Phe7-Phe8 structure of the peptide is fully drawn, to indicate which fragment of substance P was addressed to be mimicked. The 3D superimposition of the two molecules is shown on the following page.

Mimicry of CGP-47899 and Substance P¶
In the 3D representations, we can see how effectively two phenyl groups of CGP47899 superimpose on the Phe7-Phe8 fragment of Substance P. The superimposition of the two compounds in 3D allows us to understand why CGP47899 is a good mimic of Substance P.


Case Study-3 : Hypolipemic Agents¶
Reference Set of Hypolipemic Agents¶
Clofibrate (atromide-S) has been used for the last 30 years in the treatment of arteriosclerotic diseases by lowering cholesterol and triglycerides in blood. A great variety of clofibrate analogs have been studied showing that a second benzene ring enhances potency; treloxinate is one example.


articles
Treloxinate and Related Hypolipidemic 12H-Dibenzo(d,g)(1,3)Dioxocin-6-Carboxylate Derivatives Grisar JM, Parker RA, Kariya T, Blohm TR, Fleming RW, Petrow V, Wenstrup DL and Johnson RG J. Med. Chem. 15 1972
book
Witiak DT, Newman HAI and Feller DR In Medicinal Research Series, Vol7 Dekker 1977
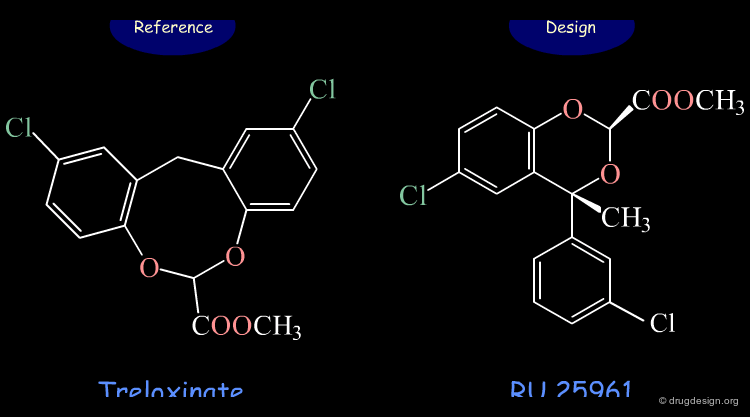
Design of a New Hypolipemic Agent¶
RU 25961 is a new hypolipemic agent that was designed on the basis of its 3D similarity with treloxinate.

articles
Synthese et Etude de l'Activite Hypolipemiante d'Acides et d'Esters Phenyl-4 /4H/ Benzodioxine-1,3 Carboxyliques-2 Humbert D, Dagnaux M, Cohen NC, Fournex R and Clemence F Eur. J. Med. Chem. 18 1983
book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
N.C. Cohen ACS-CSJ Symposium on Computer Assisted Drug Design American Chemical Society, Washington,DC; ACS Symposium Series Vol.112 1979
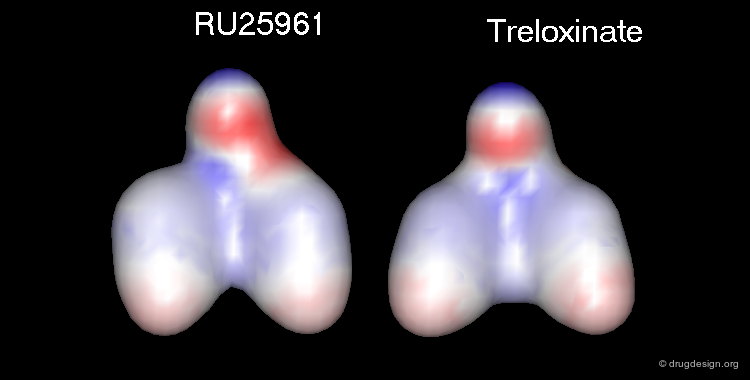
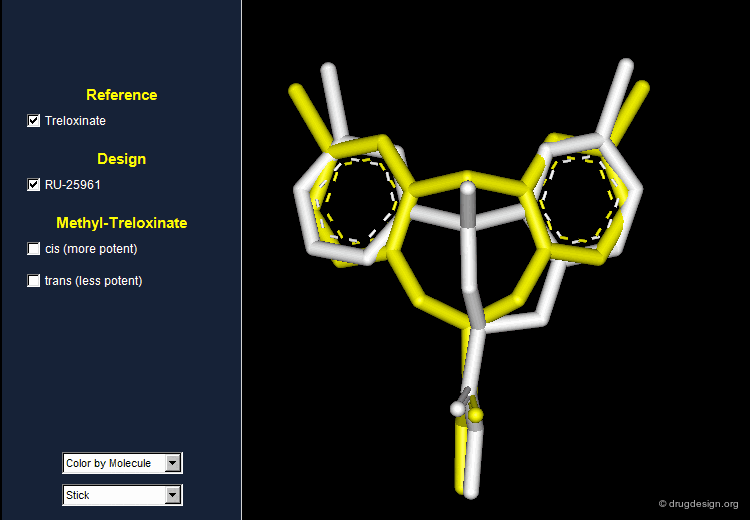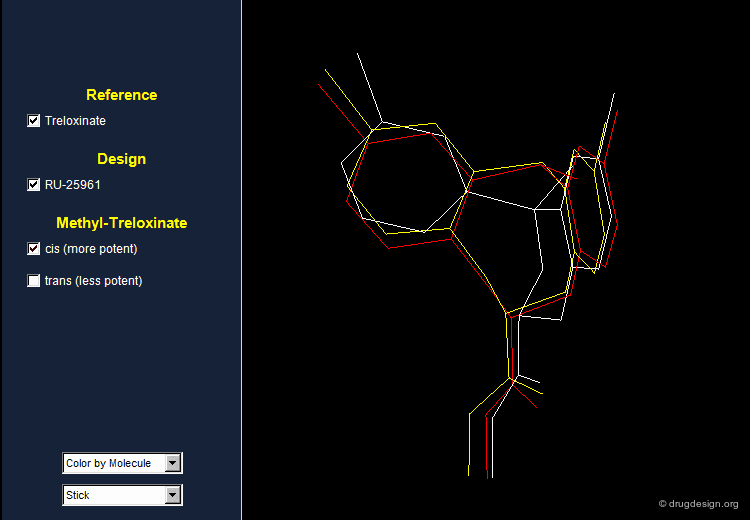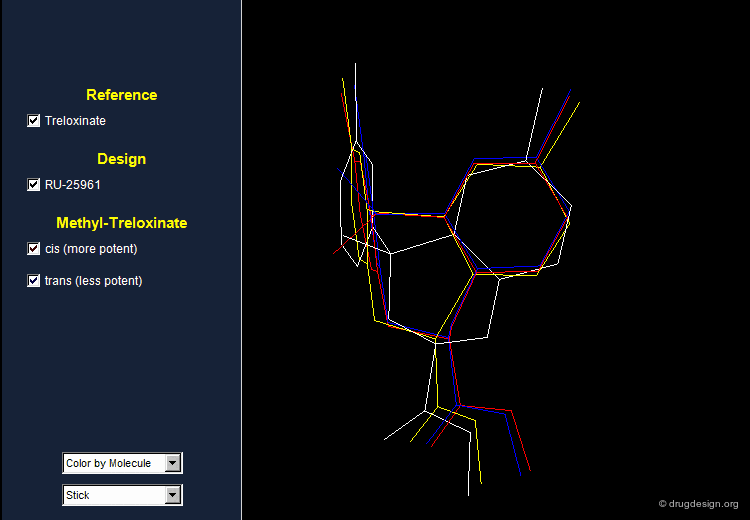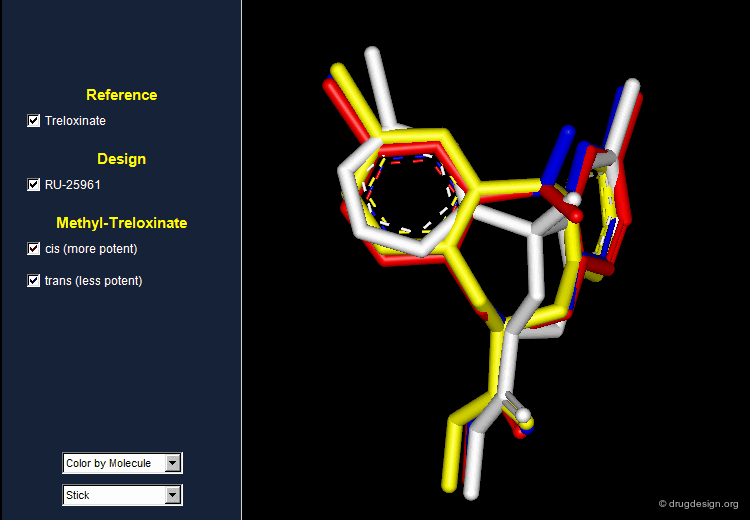
RU 25961 is a 3D Mimic of Treloxinate¶
Although having entirely different 2D chemical structures, RU 25961 and treloxinate mimic each other. Not only are the shapes of the two molecules similar, but they also present the same distribution of physicochemical properties in 3D.
![]()


book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
N.C. Cohen ACS-CSJ Symposium on Computer Assisted Drug Design American Chemical Society, Washington,DC; ACS Symposium Series Vol.112 1979
Browser of Hypolipemic Agents¶

Methyl Treloxinate¶
Treloxinate analog compounds were synthesized to optimize the series. Methyl-treloxinates were prepared by introducing the methyl substituent on the methylene bridge of the eight-membered ring. This molecule exists as a mixture of "cis" and "trans" isomers depending on the relative stereochemistry of the two substituents of the eight-membered ring. The cis isomer was shown to be more active and the trans isomer less active than the reference treloxinate compound.
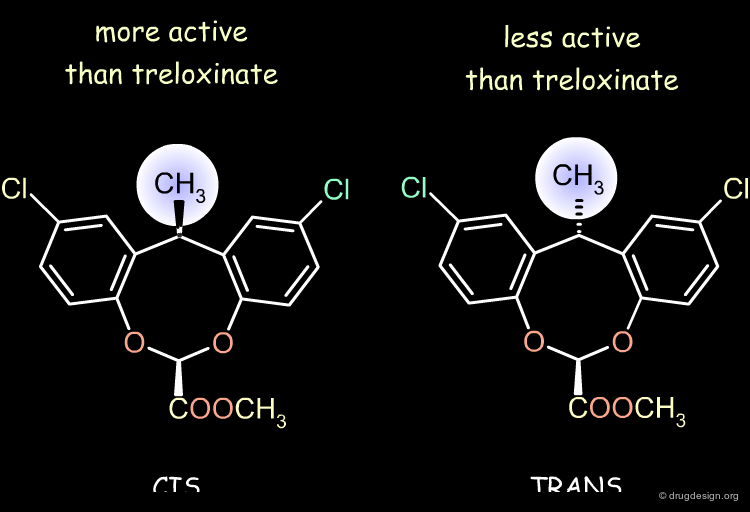
Biological Activities of Cis and Trans Isomers¶
The difference in the observed activities between the cis and trans isomers of methyl treloxinate can be understood by looking at their 3D features as compared to RU-25961. The mimicry between treloxinate and RU-25961 is more pronounced with the cis isomer, as indicated by the location of the additional methyl substituent.

book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
N.C. Cohen ACS-CSJ Symposium on Computer Assisted Drug Design American Chemical Society, Washington,DC; ACS Symposium Series Vol.112 1979
Browser of Hypolipemic Agents¶
book
N.C. Cohen Advances in Drug Research; volume 14 Academic Press 1985
N.C. Cohen ACS-CSJ Symposium on Computer Assisted Drug Design American Chemical Society, Washington,DC; ACS Symposium Series Vol.112 1979
Case Study-4 : Polymerase-1 Inhibitors¶
Therapeutic utility of PARP-1 Inhibitors¶
Poly (ADP-ribose) Polymerase-1 (PARP-1) inhibitors can be useful in the repair of DNA damage induced by radiation during cancer therapy.

articles
Tricyclic Benzimidazoles as Potent Poly(ADP-ribose) Polymerase-1 Inhibitors Skalitzky DJ, Marakovits JT, Maegley KA, Ekker A, Yu XH, Hostomsky Z, Webber SE, Eastman BW, Almassy R, Li J, Curtin NJ, Newell DR, Calvert AH, Griffin RJ and Golding BT Journal of Medicinal Chemistry 46 2003
3-Amino Benzamide PARP-1 Inhibitor¶
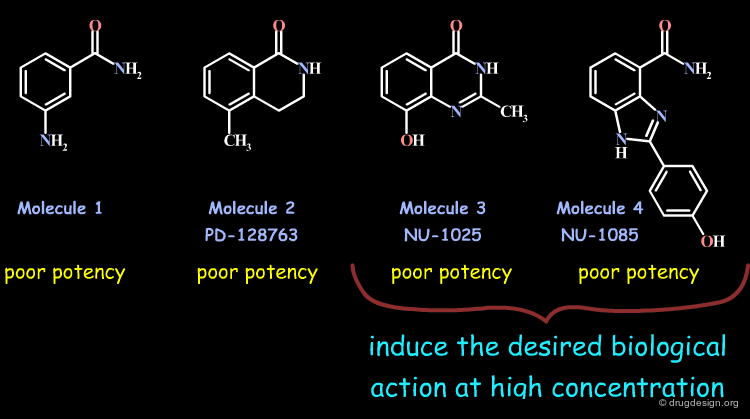
Early PARP-1 inhibitors were analogs of 3-aminobenzamide (Molecule 1) that lacked specificity and potency. Subsequent designs of constrained analogs locking the carboxamide moiety in its probable bioactive conformation (2, 3 and 4) yielded compounds with poor potency; however 3 and 4 were able to induce the desired biological properties at high concentrations.
articles
Tricyclic Benzimidazoles as Potent Poly(ADP-ribose) Polymerase-1 Inhibitors Skalitzky DJ, Marakovits JT, Maegley KA, Ekker A, Yu XH, Hostomsky Z, Webber SE, Eastman BW, Almassy R, Li J, Curtin NJ, Newell DR, Calvert AH, Griffin RJ and Golding BT Journal of Medicinal Chemistry 46 2003
Design with Carboxamide Geometry Locked¶
The design of tricyclic analogs B and C of compound A was considered. Further analyses led to the rejection of B because of potential problems in both synthesis and stability. The tricyclic molecule C emerged as a promising candidate that could mimic the initial reference compound 4 (NU-1085) well.

3D Mimicry between Structure C and NU-1085¶
The following view illustrates the mimicry between the candidate structure C and the reference compound 4 (NU-1085).

articles
Tricyclic Benzimidazoles as Potent Poly(ADP-ribose) Polymerase-1 Inhibitors Skalitzky DJ, Marakovits JT, Maegley KA, Ekker A, Yu XH, Hostomsky Z, Webber SE, Eastman BW, Almassy R, Li J, Curtin NJ, Newell DR, Calvert AH, Griffin RJ and Golding BT Journal of Medicinal Chemistry 46 2003
Synthesis of the Designed Tricyclic Compounds¶
Prototype candidates were synthesized and led to the discovery of potent inhibitors. SAR indicate that the carboxamide hydrogen is essential; its replacement by a methyl group leads to a complete loss of binding affinity. Structure 22 represents a 25-fold increase in efficacy when compared to the reference 4 (NU-1085) molecule and shows the best biological profile in cellular assays.

articles
Tricyclic Benzimidazoles as Potent Poly(ADP-ribose) Polymerase-1 Inhibitors Skalitzky DJ, Marakovits JT, Maegley KA, Ekker A, Yu XH, Hostomsky Z, Webber SE, Eastman BW, Almassy R, Li J, Curtin NJ, Newell DR, Calvert AH, Griffin RJ and Golding BT Journal of Medicinal Chemistry 46 2003
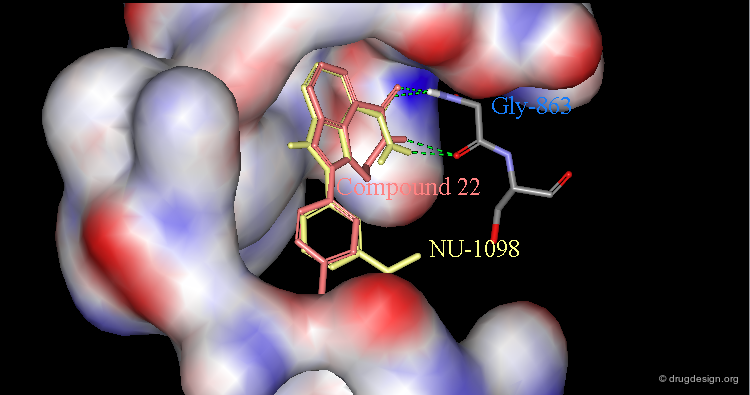
Validation of the Concept by X-Ray Crystallography¶
The design concept was validated by X-ray crystallographic analyses of complexes with PARP-1. Here are visualized compound 22 and NU-1098 (the methoxy-analog of 4) bound to the catalytic site of the protein. In particular it shows the bound geometry of the amino-benzamide pharmacophore and also explains the importance of the carboxamide hydrogen that is liganded to Gly-863.
articles
Resistance-Modifying Agents. 9.(1) Synthesis and Biological Properties of Benzimidazole Inhibitors of the DNA Repair Enzyme Poly(Adp-Ribose) Polymerase White AW, Almassy R, Calvert AH, Curtin NJ, Griffin RJ, Hostomsky Z, Maegley K, Newell DR, Srinivasan S and Golding BT Journal of Medicinal Chemistry 43 2000
Browser of PARP-1 Inhibitors¶

articles
Tricyclic Benzimidazoles as Potent Poly(ADP-ribose) Polymerase-1 Inhibitors Skalitzky DJ, Marakovits JT, Maegley KA, Ekker A, Yu XH, Hostomsky Z, Webber SE, Eastman BW, Almassy R, Li J, Curtin NJ, Newell DR, Calvert AH, Griffin RJ and Golding BT Journal of Medicinal Chemistry 46 2003
Case Study-5 : Angiotensin-II Antagonists¶
Antagonists of Angiotensin-II Receptors¶
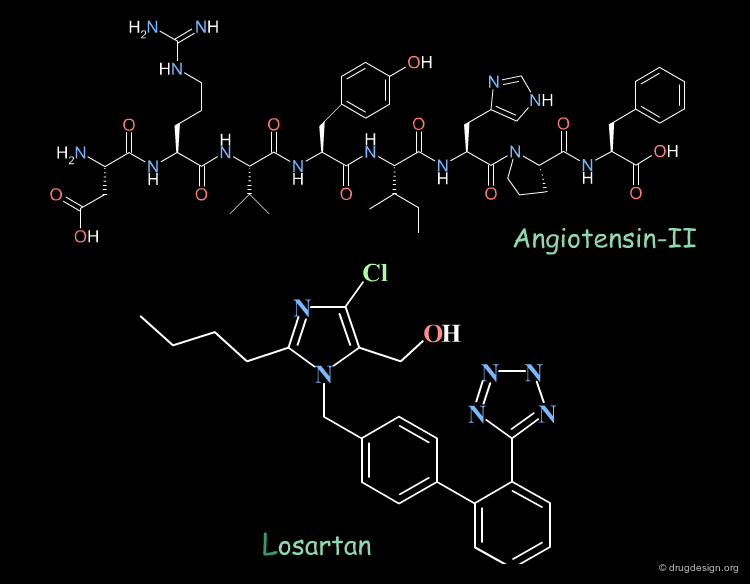
Angiotensin-II is the octapeptide substrate of Angiotensin-II receptors. Losartan is a potent angiotensin-II antagonist, a new class of anti-hypertensive agents. Its scaffold was discovered by traditional screening and further optimized for potency and selectivity. An interesting question is whether Losartan and Angiotensin-II have 3D features in common; in other words is Losartan a 3D mimic of Angiotensin-II ? Arguments in favor of this hypothesis are presented on the following pages.
articles
An Effort to Understand the Molecular Basis of Hypertension Through the Study of Conformational Analysis of Losartan and Sarmesin Using a Combination of Nuclear Magnetic Resonance Spectroscopy and Theoretical Calculations Mavromoustakos T, Kolocouris A, Zervou M, Roumelioti P, Matsoukas J and Weisemann R J. Med. Chem. 42 1999
Losartan as a Mimic of Angiotensin-II (1/5)¶
The tetrazole of Losartan can mimic the C-terminal carboxylate of the peptidic Angiotensin-II.

articles
Use of Molecular Modeling to Study Superimposition of Angiotensin II eith its Non-peptide Receptor Antagonists Mavromoustakos T, Theodoropoulou E, Matsoukas J, Moore G and Smith J Epitheorese Klinikes Farmakologias Kai Farmakokinetikes International Edition. 8 1994
Structure Elucidation and Conformational Properties of Eprosartan a Non Peptide Angiotensin II AT(1) Antagonist Zoumpoulakis P, Grdadolnik SG, Matsoukas J and Mavromoustakos T J. Pharm. Biomed. Anal. 28 2002
Losartan as a Mimic of Angiotensin-II (2/5)¶
The imidazole ring of Losartan is in the vicinity of the histidine ring of Angiotensin-II.

articles
Use of Molecular Modeling to Study Superimposition of Angiotensin II eith its Non-peptide Receptor Antagonists Mavromoustakos T, Theodoropoulou E, Matsoukas J, Moore G and Smith J Epitheorese Klinikes Farmakologias Kai Farmakokinetikes International Edition. 8 1994
Structure Elucidation and Conformational Properties of Eprosartan a Non Peptide Angiotensin II AT(1) Antagonist Zoumpoulakis P, Grdadolnik SG, Matsoukas J and Mavromoustakos T J. Pharm. Biomed. Anal. 28 2002
Losartan as a Mimic of Angiotensin-II (3/5)¶
The hydroxyl-methyl group of Losartan is close to the hydroxyl group of the tyrosine of Angiotensin-II.

articles
Use of Molecular Modeling to Study Superimposition of Angiotensin II eith its Non-peptide Receptor Antagonists Mavromoustakos T, Theodoropoulou E, Matsoukas J, Moore G and Smith J Epitheorese Klinikes Farmakologias Kai Farmakokinetikes International Edition. 8 1994
Structure Elucidation and Conformational Properties of Eprosartan a Non Peptide Angiotensin II AT(1) Antagonist Zoumpoulakis P, Grdadolnik SG, Matsoukas J and Mavromoustakos T J. Pharm. Biomed. Anal. 28 2002
Losartan as a Mimic of Angiotensin-II (4/5)¶
The hydrophobic butyl chain of Losartan mimics the alkyl chain of the isoleucine aminoacid of Angiotensin-II.

articles
Use of Molecular Modeling to Study Superimposition of Angiotensin II eith its Non-peptide Receptor Antagonists Mavromoustakos T, Theodoropoulou E, Matsoukas J, Moore G and Smith J Epitheorese Klinikes Farmakologias Kai Farmakokinetikes International Edition. 8 1994
Structure Elucidation and Conformational Properties of Eprosartan a Non Peptide Angiotensin II AT(1) Antagonist Zoumpoulakis P, Grdadolnik SG, Matsoukas J and Mavromoustakos T J. Pharm. Biomed. Anal. 28 2002
Losartan as a Mimic of Angiotensin-II (5/5)¶
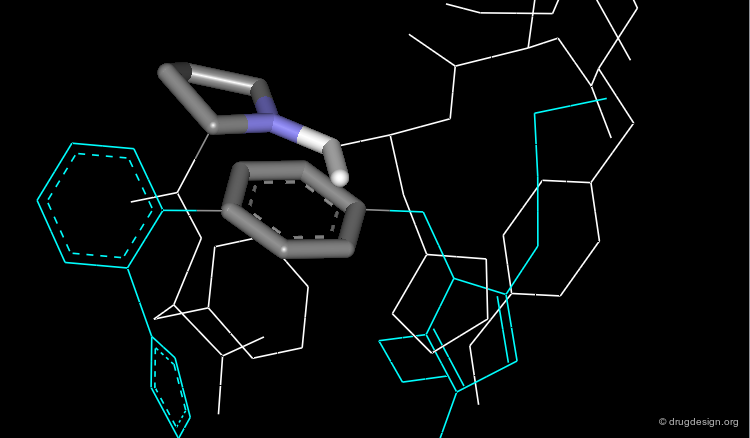
The biphenyl template of Losartan is in close proximity with the phenyl and proline rings of Angiotensin-II that mimic a β-turn conformation.
articles
Use of Molecular Modeling to Study Superimposition of Angiotensin II eith its Non-peptide Receptor Antagonists Mavromoustakos T, Theodoropoulou E, Matsoukas J, Moore G and Smith J Epitheorese Klinikes Farmakologias Kai Farmakokinetikes International Edition. 8 1994
Structure Elucidation and Conformational Properties of Eprosartan a Non Peptide Angiotensin II AT(1) Antagonist Zoumpoulakis P, Grdadolnik SG, Matsoukas J and Mavromoustakos T J. Pharm. Biomed. Anal. 28 2002
Browser of Angiotensin-II Antagonists¶

articles
Derivation of a 3D Pharmacophore Model for the Angiotensin-II Site One Receptor Prendergast K, Adams K, Greenlee WJ, Nachbar RB, Patchett AA and Underwood DJ J. Comput. Aided Mol. Des. 8 1994
Nonpeptide Angiotensin II Receptor Antagonists: the Next Generation in Antihypertensive Therapy Wexler RR, Greenlee WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD and Timmermans PB J Med Chem. 39 1996
Case Study-6 : Cholecystokinin Receptor Ligands¶
Design of Cholecystokinin Receptor Ligands¶
MK-329 was found to be a very potent and selective CCK receptor antagonist with an affinity comparable to that of the native peptide. Lorglumide, an analog of glutamic acid, was also known to have good CCK selectivity but poor potency. On the following pages we discuss how analysis of the molecular mimicry features of these two molecules led to the design and discovery of potent analogs of the poorly active Lorglumide molecule.

Pharmacophore Analysis: CCK-A Antagonists (1/3)¶
By using pharmacophore alignments, two pharmacophoric elements were found between the non-peptide antagonist MK-329 and Lorglumide.

articles
Novel Glutamic Acid Derived Cholecystokinin Receptor Ligands Freidinger RM, Whitter WL, Gould NP, Holloway MK, Chang RS and Lotti VJ J. Med. Chem. 33 1990
Pharmacophore Analysis: CCK-A Antagonists (2/3)¶
The hypothesis was that the indolecarboxamide in MK-329 is mimicked by substituting the phenyl ring of the Lorglumide.

articles
Novel Glutamic Acid Derived Cholecystokinin Receptor Ligands Freidinger RM, Whitter WL, Gould NP, Holloway MK, Chang RS and Lotti VJ J. Med. Chem. 33 1990
Pharmacophore Analysis: CCK-A Antagonists (3/3)¶
The n-pentyl group of Lorglumid mimics the hydrophobic phenyl and benzo groups of MK-329.
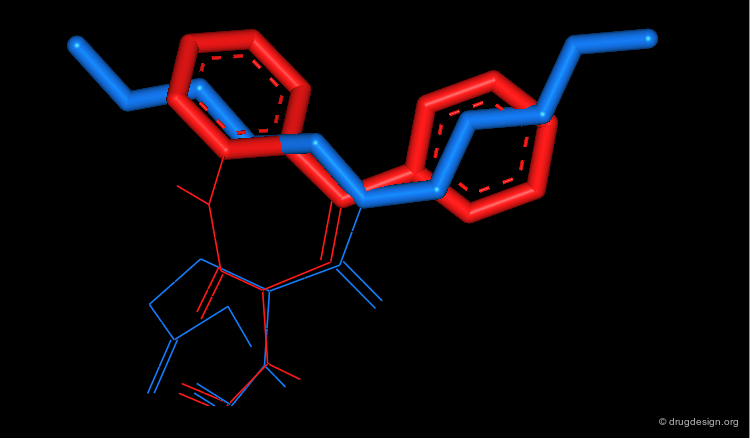
articles
Novel Glutamic Acid Derived Cholecystokinin Receptor Ligands Freidinger RM, Whitter WL, Gould NP, Holloway MK, Chang RS and Lotti VJ J. Med. Chem. 33 1990
Design of a New Lorglumide Analog¶
On the basis of a previous pharmacophore hypothesis, a new analog of Lorglumide was designed. Replacing the phenyl ring with an indole group increased the affinity for the CCK receptor by a factor of 3.
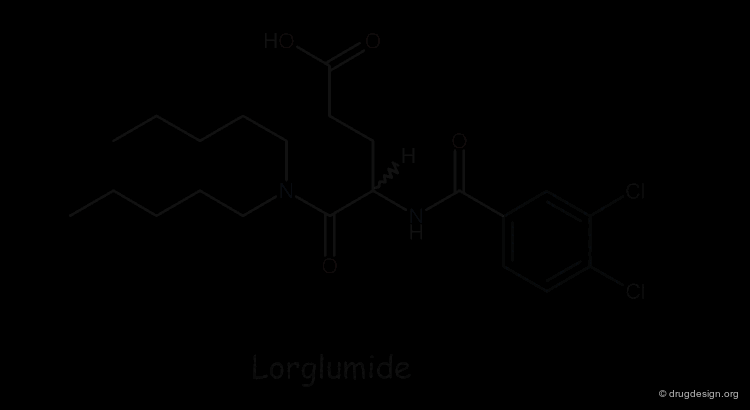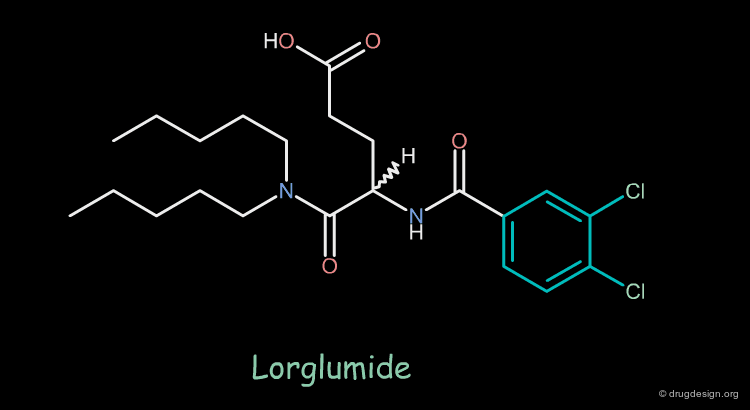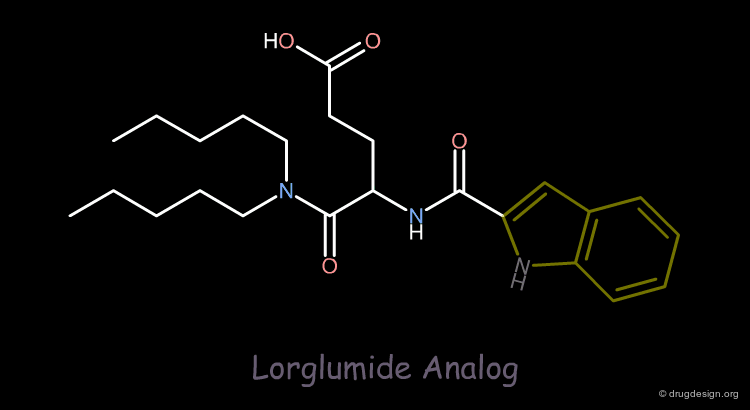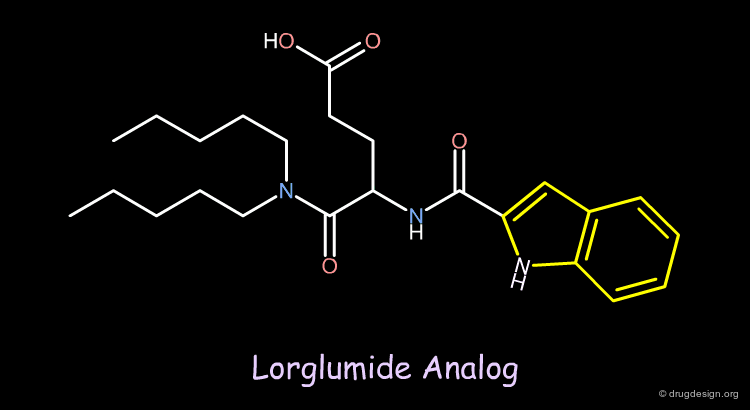

articles
Novel Glutamic Acid Derived Cholecystokinin Receptor Ligands Freidinger RM, Whitter WL, Gould NP, Holloway MK, Chang RS and Lotti VJ J. Med. Chem. 33 1990
Case Study-7 : Farnesyltransferase Inhibitors¶
Farnesyltransferase, a Target in Oncology¶
Farnesyltransferase (FTase) is a zinc metalloenzyme that catalyzes farnesylation of proteins with a carboxyl terminal sequence CAAX, where C is cysteine, A is an aliphatic amino-acid and X is methionine, serine, alanine, glutamine or cysteine. FTase is a biological target in oncology owing to its involvement in the Ras pathway. In the following pages we present how several potent and selective non-peptidic inhibitors were discovered by fully exploiting the mimicry with a reference peptide ligand.

X-ray Structure of FTase with a Tetrapeptide¶
A crystal structure of FTase with the Ac-Cys-Val-Ile-Selenomethionine-OH tetrapeptide shows that the ligand adopts an extended conformation in which the cysteine thiol group coordinates to the catalytic zinc ion. The Cys-Val-Ile-Met peptide inhibits FTase in vitro (IC50 = 340 nM).
articles
Crystal Structure of Farnesyl Protein Trasnferase Complexed with a Caax Peptide and Farnesyl Diphosphate Analogue Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS and Weber PC Biochemistry 37 1998
Binding Interactions of CAAX Substrate for FTase¶
The interactions of the Ac-Cys-Val-Ile-Met(Se)-OH ligand are schematically represented in the diagram below. This structure can be a starting point for the design of peptidomimetics and non-peptide inhibitors of the FTase.

4-Aminobenzoic Spacer to Replace Val-Ile Dipeptide¶
The following modifications of the initial tetrapeptide were made: (1) a rigid 4-aminobenzoic spacer to replace the Val-Ile dipeptide fragment and (2) the replacement of the cysteine amide bond by a methyleneamino group. The resulting molecule 2 proved to inhibit FTase with an IC50 of 267 nM.

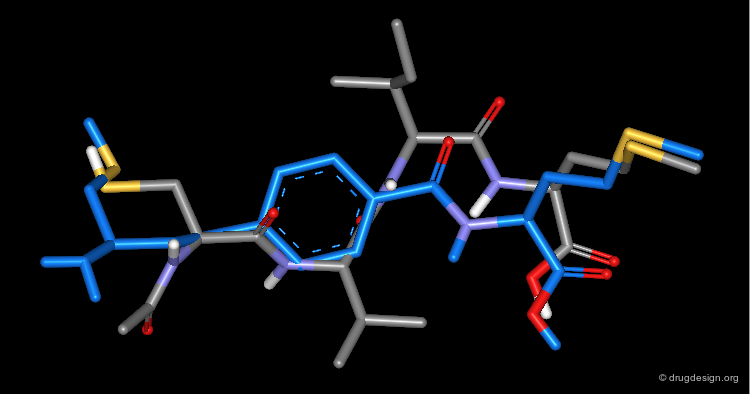
Superposition with X-Ray Structure of Initial Tripeptide¶
The view shown here visualizes the superposition of compound 2 (in blue) with the crystal structure of the initial tetrapeptide and illustrates the underlying design idea.
The Simple Aromatic Central Ring is not Sufficient¶
The 4-aminobenzoic moiety mimics the corresponding Val-Ile backbone however the simple aromatic ring does not reach the volume occupied by the side-chains of the dipeptide. Substituted analogs were designed to improve the overlap with the reference peptide.

Analogs with Significantly Enhanced Potency¶
The substitution of the phenyl at the 2-position significantly enhances potency. Not only a phenyl group but other substituents such as 2-thienyl or 1-naphthyl also yield potent inhibitors. The methyl ester of the initial phenyl derivative showed significant potency in inhibiting Ras farnesylation in whole cells.

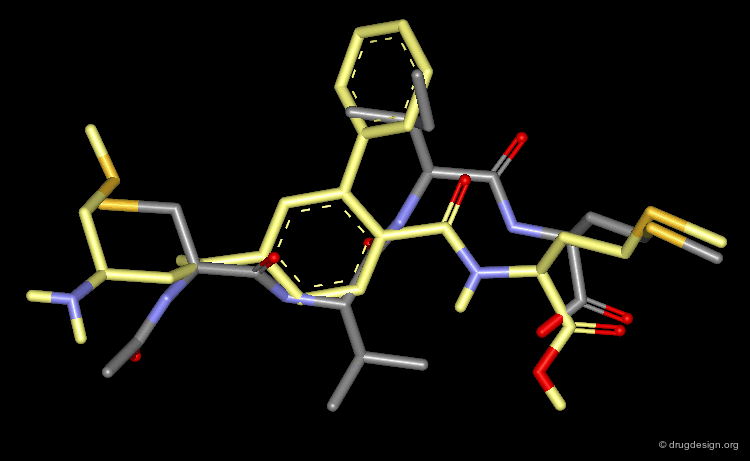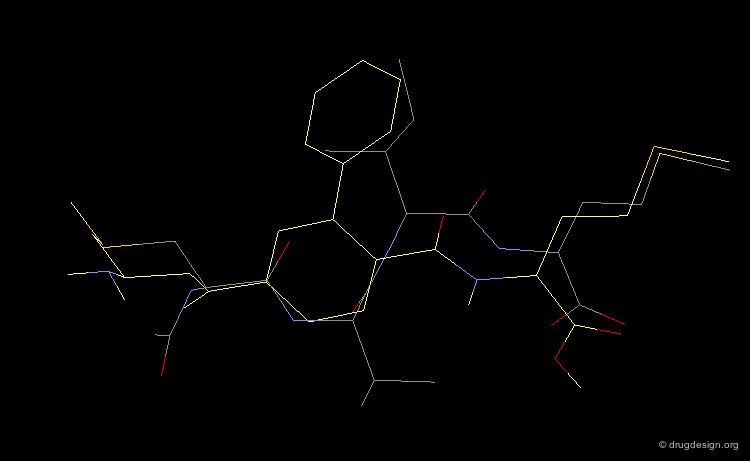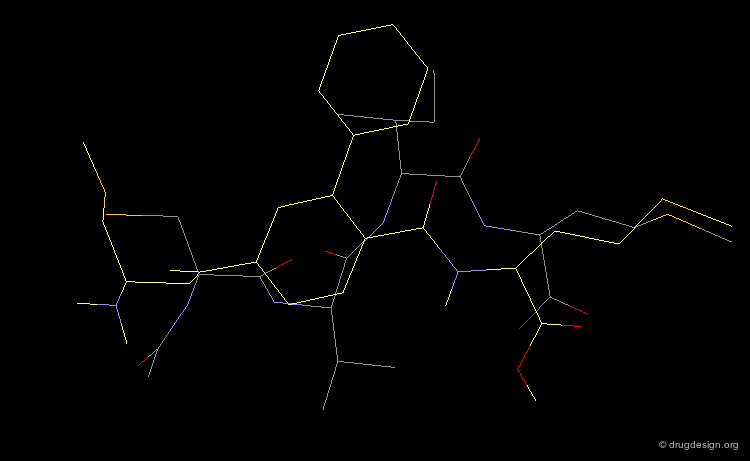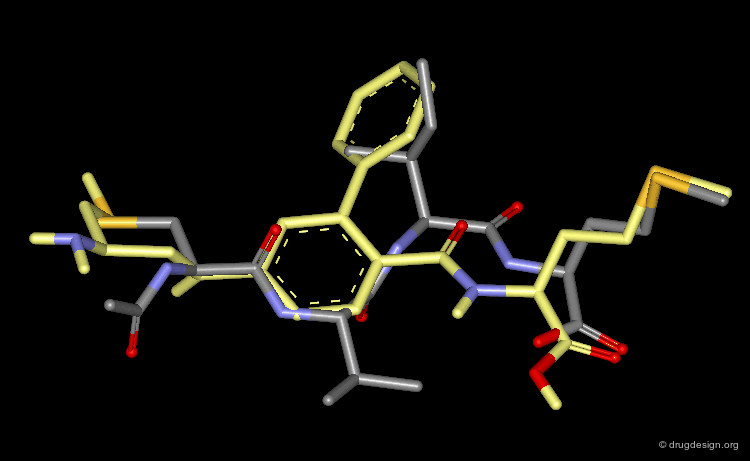
3D Mimicry of FTI-276 and Reference Tetrapeptide¶
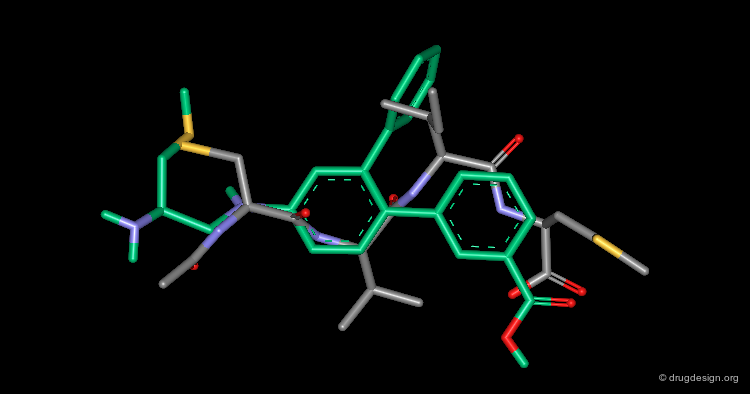
This view shows the 3D mimicry of the 2-phenyl-4-aminobenzoic acid analog (FTI-276, in yellow) with the crystal structure of the reference tetrapeptide.
Docking of FTI-276 and Reference Tetrapeptide¶
The tetrapeptide and the FTI-276 inhibitor are shown docked in the catalytic site of the enzyme. Both ligands possess a hydrophobic fragment (isoleucine/phenyl) that penetrates deep into a hydrophobic pocket of the enzyme. Note that the absence of significant interactions with the amide fragment located between the Val and Ile residues justifies the design of hydrophobic spacers in this region.

Terphenyl to Replace the Central Val-Ile Dipeptide¶
A similar design considering a terphenyl template also proved to be successful. For example FTI-289 is a very potent inhibitor of FTase, with an IC50 of 20 nM. Moreover, FTI-2297 (IC50 = 380 nM) demonstrates the possibility of discovering inhibitors with no chiral centers.

Alignment of FTI-289 with Cys-Val-Ile-Met¶
The following view visualizes the alignment of FTI-289 with the X-ray structure of the Cys-Val-Ile-Met tetrapeptide.
Potent and Selective Farnesyltransferase Inhibitor¶
Very recently Abbott discovered fully non-peptidic and achiral inhibitors. For example Abbott-21 is a potent inhibitor of FTase (IC50 = 0.39 nM), FTase/GTase selective (>300-fold) and cellularly active (<80 nM).
articles
Novel and selective imidazole-containing biphenyl inhibitors of protein farnesyltransferase Curtin ML, Florjancic AS, Cohen J, Gu WZ, Frost DJ, Muchmore SW and Sham HL Bioorganic Medicinal Chemistry Letters 13 2003
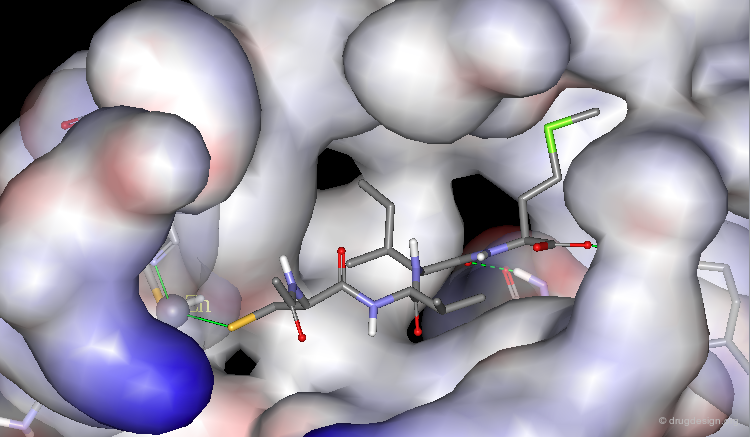
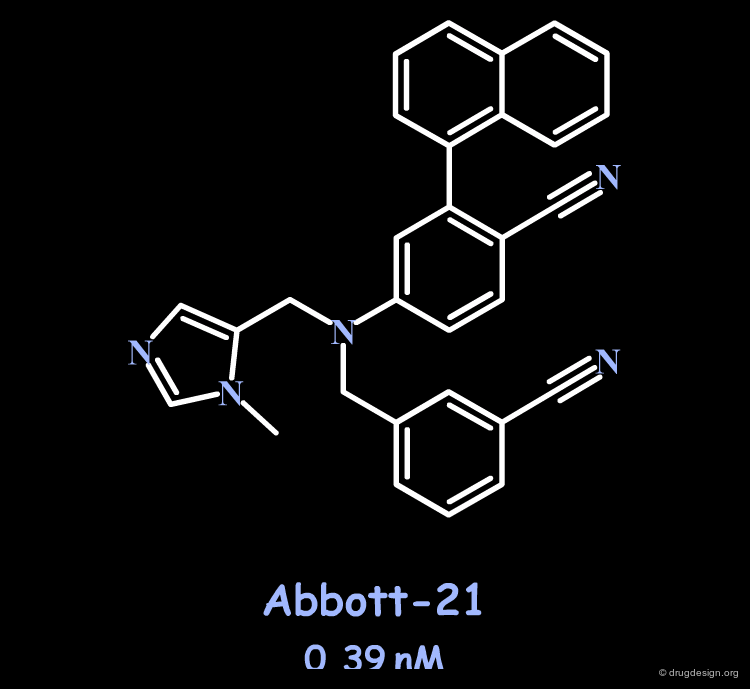
X-ray of Abbott-21 bound to Farnesyltransferase¶
The X-ray crystal structure of the Abbott-21 inhibitor bound to the farnesyltransferase proved that it fulfilled the design criteria presented in the preceding pages. In addition, this confirms that enhanced activities and selectivity can be achieved by mimicking the Ile - and not Val - of the reference tetrapeptide. Note the two hydrogen bonds created with the cyano fragments and the coordination with the zinc atom.
Browser of Farnesyltransferase Inhibitors¶

Case Study-8 : Antagonists of the Mdm2-p53 Interaction¶
Antagonists of the Mdm2-p53 Interaction¶
The Mdm2 and p53 genes normally form a negative feedback loop in cells to monitor and regulate p53 ability to limit growth. p53 is an attractive therapeutic target in oncology because its tumor suppressor activity can be stimulated to eradicate tumor cells. A recent approach to stimulate p53 activity is based on the inhibition of the p53-Mdm2 interaction with synthetic molecules. Peptide and non-peptide inhibitors were found by molecular mimicry, as explained on the following pages.

Mdm2 Bound to p53 Transactivation Domain (1/4)¶
The crystal structure of the 109-residue amino terminal domain of the Mdm2 protein bound to a 15-residue peptide from the transactivation domain of p53 reveals the nature of the interaction.

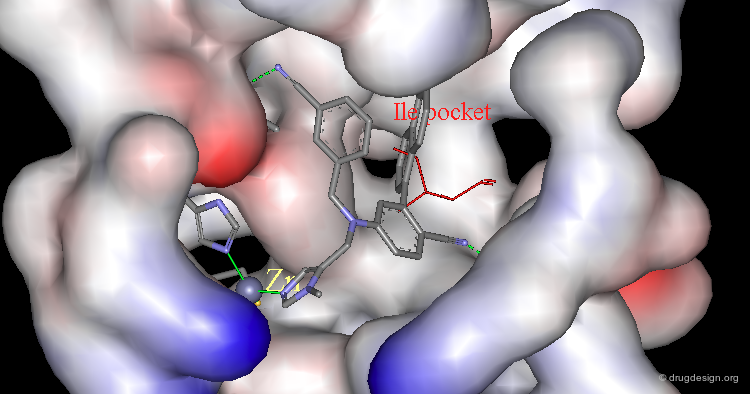
Mdm2 Bound to p53 Transactivation Domain (2/4)¶
In particular the presence of a deep hydrophobic cleft on the surface of Mdm2 was found.
Mdm2 Bound to p53 Transactivation Domain (3/4)¶
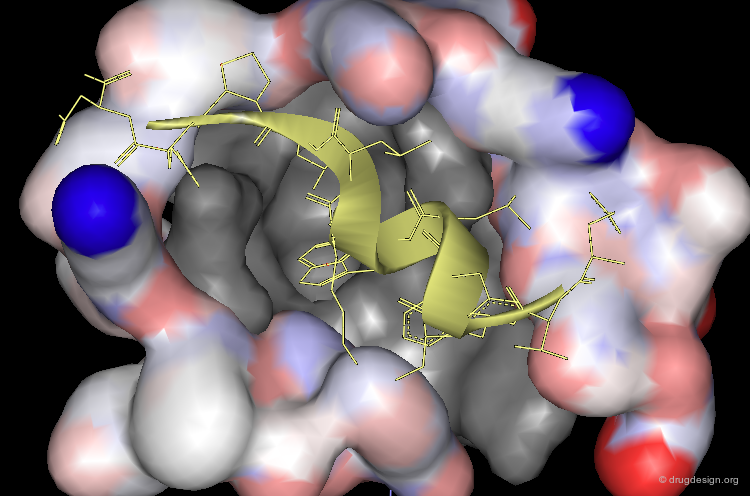
This cleft accommodates the p53 peptide, which adopts an amphipathic α-helix conformation.
Mdm2 Bound to p53 Transactivation Domain (4/4)¶
Moreover, the interaction is dominated by complementary geometric features involving a triad of p53 amino-acids Phe-19, Trp-23 and Leu-26 that penetrate deep into the Mdm2 cleft.

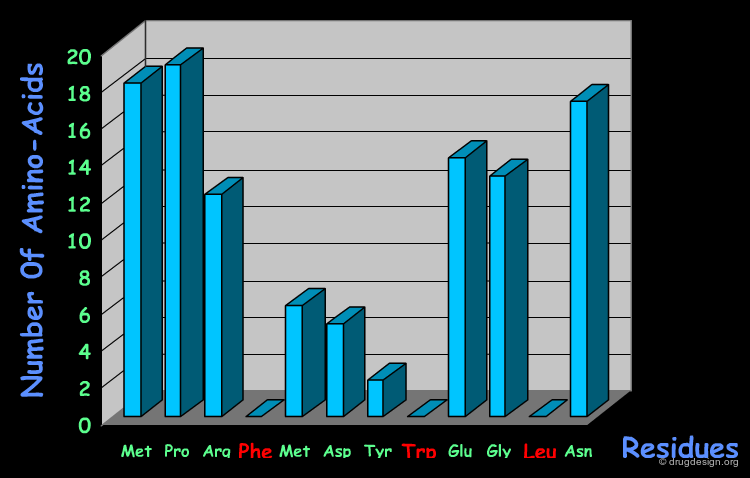
Systematic SAR Studies¶
A systematic SAR study made on a 12-mer reference peptide antagonist, and replacing each amino-acid individually by each of the other 19 possible amino-acids, revealed the importance of the three amino-acids Phe-19, Trp-23 and Leu-26 that are "not permitted" replacements.
articles
Molecular Characterization of the hdm2-p53 Interaction Bottger A, Bottger V, Garcia-Echeverria C, Chene P, Hochkeppel HK, Sampson W, Ang K, Howard SF, Picksley SM and Lane DP Journal of Molecular Biology 269 1997
Computational Alanine Scanning To Probe Protein-Protein Interactions: A Novel Approach To Evaluate Binding Free Energies Massova I and Kollman PA Journal of the American Chemical Society 121 1999
Contribution of the Amino-Acids to the Binding¶
Advanced theoretical calculations (computational alanine-scanning) served to estimate the contribution of each amino-acid of a small peptide antagonist derived from the p53 transactivation domain.

3D Structure of the Pharmacophore¶
The structural studies therefore reveal the importance of the three amino-acids of the p53 protein: Phe-19, Trp-23 and Leu-26. They define a pharmacophore that can serve as a starting point for the design of new inhibitors.

The Novartis 5 nM Peptide-Like Antagonist¶
The Novartis group succeeded in discovering potent peptide antagonists, which started by a phage display study that led to the identification of an initial lead represented by peptide 1.

articles
Discovery of Potent Antagonists of the Interaction between Human Double Minute 2 and Tumor Suppressor p53 Garcia-Echeverria C, Chene P, Blommers MJJ and Furet P Journal of Medicinal Chemistry 43 2000
Peptide 2 Designed to Stabilize Helical Conformations¶
In the second phase, residues that do not interact with Mdm2 (e.g. Asp and Gly) were modified in order to stabilize the α-helix conformation. C-α disubstituted amino-acids are known to stabilize α and 310-helices. Peptide 2 was developed along this line and showed an IC50 of 2.2 µM.

Peptide 3 Designed for a Salt Bridge with a Tyrosine¶
The proximity of the p-53 peptide residue Tyr-22 and the Mdm2 Lys-94 side chain led to the idea of replacing the tyrosine by a group that would form a salt bridge with the amino group of Lys-94; this led to peptide 3 with an IC50 of 314 nM.

Filling Empty Space Identified by Modeling¶
Modeling analyses revealed the existence of an empty space located in the direction of the 6-position of the indole ring. This space corresponded to the volume of a methyl group or a chlorine atom. Both molecules showed a major increase in binding affinity (e.g. IC50 of 5 nM for peptide 4).

Problems with the Peptide-Based Antagonists¶
Further efforts to develop the discovered peptide inhibitors did not succeed. Novartis came to the conclusion that they were "by no means drugs". The peptide-like molecules were shown to have very poor activity in cellular assays. At this stage, looking for non-peptidic inhibitors using a de novo design strategy might be the way to discover useful compounds with good cellular activities.

The Bicyclo [2.2.1]-Heptane Scaffold¶
A de novo approach was considered with the bicyclo [2.2.1]-heptane scaffold shown below. This is the first report of a de novo attempt to design non-peptidic molecules to antagonize the Mdm2-p53 interaction.

articles
Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53 Garcia-Echeverria C, Chene P, Blommers MJ and Furet P J. Med. Chem 43 2000
Computational Alanine Scanning To Probe Protein-Protein Interactions: A Novel Approach To Evaluate Binding Free Energies Massova I, Kollman PA. J. Am. Chem. Soc. 121 1999
Structure-Based Design of Spiro-oxindoles as Potent, Specific Small-Molecule Inhibitors of the MDM2-p53 Interaction Ke Ding, Yipin Lu, Zaneta Nikolovska-Coleska, Guoping Wang, Su Qiu, Sanjeev Shangary, Wei Gao, Dongguang Qin, Jeanne Stuckey, Krzysztof Krajewski, Peter P. Roller, and Shaomeng Wang J. Med. Chem. 49 2006
Discovery and Cocrystal Structure of Benzodiazepinedione HDM2 Antagonists That Activate p53 in Cells Bruce L. Grasberger, Tianbao Lu, Carsten Schubert, Daniel J. Parks, Theodore E. Carver, Holly K. Koblish, Maxwell D. Cummings et al. J. Med. Chem. 48 2005
Novel 1,4-benzodiazepine-2,5-diones as Hdm2 antagonists with improved cellular activity Leonard K, Marugan JJ, Raboisson P, Calvo R, Gushue JM, Koblish HK, Lattanze J, Zhao S, Cummings MD, Player MR, Maroney AC, Lu T. Bioorg Med Chem Lett 16(13) 2006
Designed Scaffold Aligned with the Pharmacophore¶
The alignment of the molecular geometry of the most active bicyclo [2.2.1]-heptane derivative with the p53 pharmacophore indicates that the design could be improved. The molecule has poor biological activities in-vitro but was shown to be capable of inducing apoptosis on some tumor cell lines.

ADDITIONAL CASE STUDIES¶
Additional Case Studies¶
Copyright © 2024 drugdesign.org