Introduction to Protein-Ligand Binding¶
Info
The analytical process and principles underlying a structure-based drug design approach are presented and discussed. Methods for analyzing binding are presented and discussed.
Number of Pages: 75 (±1 hours read)
Last Modified: January 2004
Prerequisites: Principles of Rational Drug Design
Introduction¶
Receptor-Based Drug Design¶
The aim of receptor structure-based drug design is to create new molecules with high affinity and with macromolecular receptors whose 3D structure is known. The molecules are conceived based on their 3D complementary features with the macromolecular target. In addition, one can exploit the molecular interactions to strengthen the binding of the current prototype (inhibitor, agonist or antagonist) into the active site of the receptor.

Macromolecular Targets¶
Macromolecular targets are selected on the basis of their role in the biological system involved for the designated disease. Specific biological pathways can either be blocked or magnified to facilitate the desired therapeutic action. The most common targets of drugs are proteins.

Mechanism of Action of Drugs¶
Drugs act as inhibitors of enzymatic proteins by inhibiting the generation or degradation of molecular components of the cell (e.g. inhibition of enzymes which are essential for bacterial survival by antibiotics). They can act either as receptor agonists by mimicking the effect of an endogenous chemical signal (e.g. nicotine agonists in the treatment of Parkinson disease) or as antagonists, which blocks the signal (e.g. Substance P antagonists for the release of pain).

Drug Targets¶
Drugs act on antibodies, structural proteins and ion channels (e.g. local anesthetics). Nucleic acids are also frequently used as targets. The interaction can be either direct (e.g. intercalation agents in the treatment of cancer) or indirect through proteins which regulate the replication and expression of genes. Lipids and saccharides also act as drug targets.

Contribution of Recombinant Technologies¶
The ability to reproduce significant recombinant proteins has had a great impact on the screening process and has facilitated the structure determination of many of these biologically relevant targets.

Operational Strategy: Docking¶
Docking is an energy-based operation for exploring the binding modes of two interacting molecules. A docking procedure is used as a guide to identify the preferred orientation of a ligand interacting with a macromolecule. The ligand can accommodate small conformational changes to avoid steric repulsions and produce favorable interactions with the receptor.
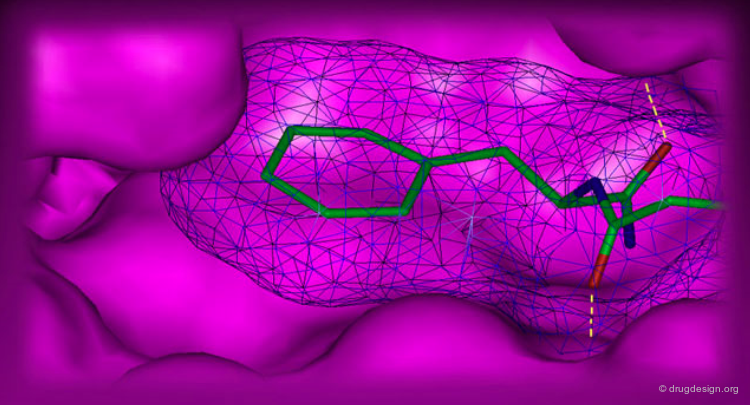
Analytical Process¶
The Analytical Process¶
Receptor-based drug design proceeds in three steps: gathering all relevant data (data collection), in-depth analyses of the information (analysis) and the design of new molecules as a result of the analysis (design).



Data Collection: X-Ray Crystallography¶
The most current method to determine the 3D structure of macromolecules is by using X-ray crystallography. This technique requires the isolation, purification and crystallization of the protein, which is not always feasible. The results are usually deposited in the Brookhaven Protein Databank, which has more than 20 thousands structures.

articles
The Protein Data Bank: A Computer-Based Archival File for Macromolecular Structures Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T and Tasumi M J. Mol. Biol. 112 1977
The Protein Data Bank Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE Nucleic Acids Research 28 2000
Automation of X-ray crystallography Abola E, Kuhn P, Earnest T and Stevens RC Nat. Struct. Biol. 7(Suppl) 2000
High-Throughput Crystallography for Lead Discovery in Drug Design Blundell TL, Jhoti H and Abell C Nature Rev. Drug Discovery 1 2002
Protein Crystallography and Computer Graphics - Toward Rational Drug Design Hol WGJ Angew Chem. Int. Ed. Engl. 25 1986
Determination of Three-Dimensional Structures of Proteins and Nucleic Acids in Solution by Nuclear Magnetic Resonance Spectroscopy Clore GM and Gronenborn AM Crit. Rev. Biochem. Mol. Biol. 24 1989
Two-, Three-, and Four-Dimensional NMR Methods for Obtaining Larger and More Precise Three-Dimensional Structures of Proteins in Solution Clore GM and Gronenborn AM Annu. Rev. Biophys. Biophys. Chem. 20 1991
NMR Spectroscopy: a Multifaceted Approach to Macromolecular Structure Ferentz AE and Wagner G Quart. Rev. Biophys. 33 2000
Introduction to the NMR of Proteins Reid DG, MacLachlan LK, Edwards AJ, Hubbard JA and Sweeney PJ Methods Mol. Biol. 60 1997
Nat. Struct. Biol. Wagner G. An Account of NMR in Structural Biology 1997 841-4 4(Suppl)
Protein Structure Determination in Solution by Nuclear Magnetic Resonance Spectroscopy Wuthrich K Science 243 1989
Modern NMR Spectroscopy of Proteins and Peptides in Solution and Its Relevance to Drug Design Zuiderweg ERP, van Doren SR, Kurochkin AV, Neubig RR and Majumdar A Perspect. Drug Discov. Design 1 1993
Protein Structure Prediction Al-Lazikani B, Jung J, Xiang Z and Honig B Curr. Opin. Chem. Biol. 5 2001
Knowledge-based Model Building of Proteins: Concepts and Examples Bajorath J, Stenkamp R and Aruffo A Protein Sci. 2 1993
Protein Structure Prediction and Structural Genomics Baker D and Sali A Science 294 2001
Knowledge-Based Prediction of Protein Structures and the Design of Novel Molecules Blundell TL, Sibanda BL, Sternberg MJ and Thornton JM Nature 326 1987
Comparative Modeling Methods: Application to the Family of the Mammalian Serine Proteases Greer J Proteins 7 1990
Knowledge-based Protein Modeling Johnson MS, Srinivasan N, Sowdhamini R and Blundell TL Crit. Rev. Biochem. Mol. Biol. 29 1994
Comparative Protein Structure Modeling of Genes and Genomes Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F and Sali A Annu. Rev. Biophys. Biomol. Struct. 29 2000
Modelling by Homology Swindells MB and Thornton JM Curr. Opin. Struct. Biol. 1 1991
book
Kenakin TP Comprehensive Medicinal Chemistry. Volume 1 Pergamon 1990
Drenth J Principles of Protein X-ray Crystallography Springer Verlag Publishinig 1999
McPherson A Chem. Struct. Approaches Ration. Drug Des CRC Press 1995
McRee DE Practical Protein Crystallography Academic Press 1990
Murray-Rust J Peptide Pharm. Ward Elsevier 1991
Rhodes G Crystallography Made Crystal Clear: A Guide for User's of Macromolecular Models Academic Press 1999
Data Collection: NMR Spectroscopy¶
NMR techniques and electron diffraction have recently been used but are limited to small proteins (up to 100-120 amino acids).

Data Collection: Homology Models¶
In the absence of a detailed 3D experimental structure, models of the macromolecular target may be used as surrogates. For example, if the primary sequence of a certain protein is known and it shares a certain degree of sequence homology with one or more proteins for which detailed structural information is available, it is possible to construct a model of the target protein using homology.

Analysis¶
Now that the raw data has been collected, one must generate additional data that can be deduced from the already existing information. This allows for the proper development of in-depth analyses that will generate new knowledge or hypotheses and the establishment of a rational link between the different components of the information. The aim of the analysis is to create a framework that integrates the knowledge that can be deduced from the structural elements that are essential for the biological activities and the stereo-chemical features of the receptor site.

articles
Molecular Recognition of Protein-Ligand Complexes: Applications to Drug Design Babine RE and Bender SL Chem. Rev. 97 1997
What Can We Learn from Molecular Recognition in Protein-Ligand Complexes for the Design of New Drugs? Bohm H-J and Klebe G Angew. Chem. Int. Ed. Engl. 35 1996
Hydrogen Bonding, Hydrophobic Interactions, and Failure of the Rigid Receptor Hypothesis Davis AM and Teague SJ Angew. Chem. Int. Ed. 38 1999
Recent Developments in Structure-Based Drug Design Klebe G J. Mol. Med. 78 2000
The Maximal Affinity of Ligands Kuntz ID, Chen K, Sharp KA and Kollman PA Proc. Natl. Acad. Sci. U.S.A. 96 1999
book
Koehler KF, Rao SN and Snyder JP Guidebook on Molecular Modeling in Drug Design Academic Press 1996
Design Phase¶
The design process can be initiated when sufficient rationales are established for the creation of molecules conforming to the desired requirements. It aims at the creation of a new molecular entity with optimal binding to the receptor site.
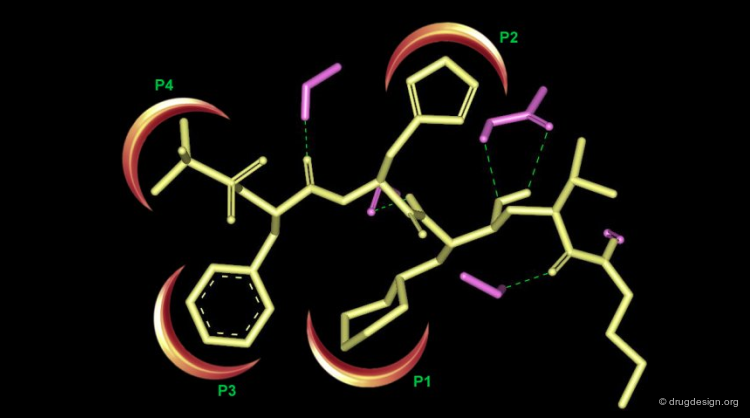
articles
What Can We Learn from Molecular Recognition in Protein-Ligand Complexes for the Design of New Drugs? Bohm H-J and Klebe G Angew. Chem. Int. Ed. Engl. 35 1996
Recent Developments in Structure-Based Drug Design Klebe G J. Mol. Med. 78 2000
book
Koehler KF, Rao SN and Snyder JP Guidebook on Molecular Modeling in Drug Design Academic Press 1996
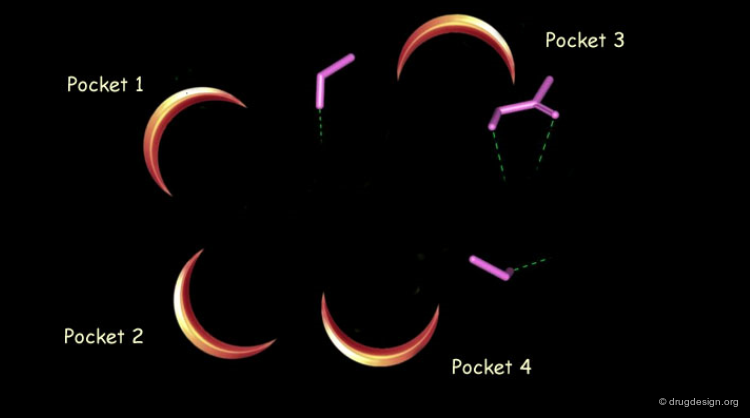
Principles of Analysis¶
Analysis of the Morphology of the Active Site¶
The shape and features of the active site are carefully analyzed. Its morphology is thoroughly inspected and broken into components such as pockets and clefts. The analysis of the morphology of an active site is greatly facilitated when a complex between interacting molecules is available.
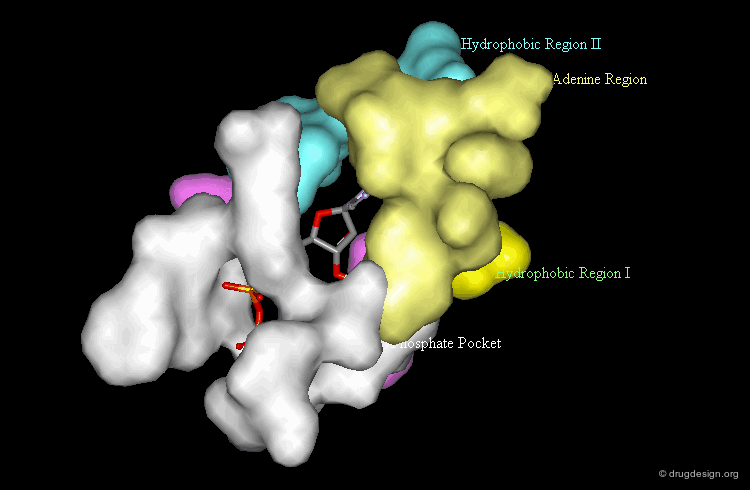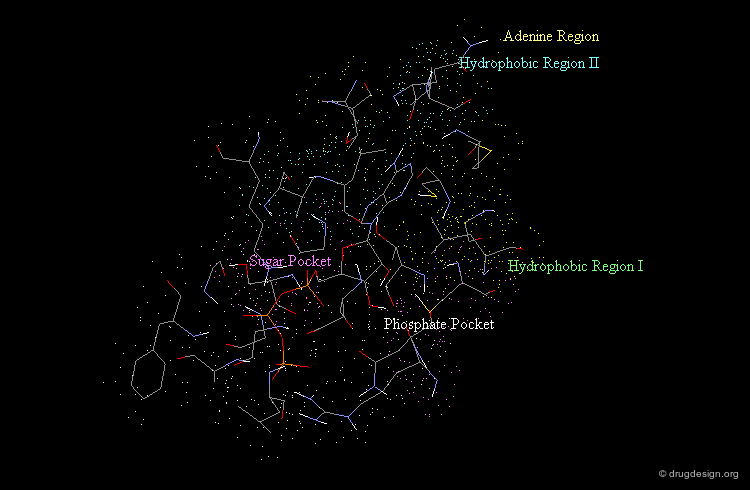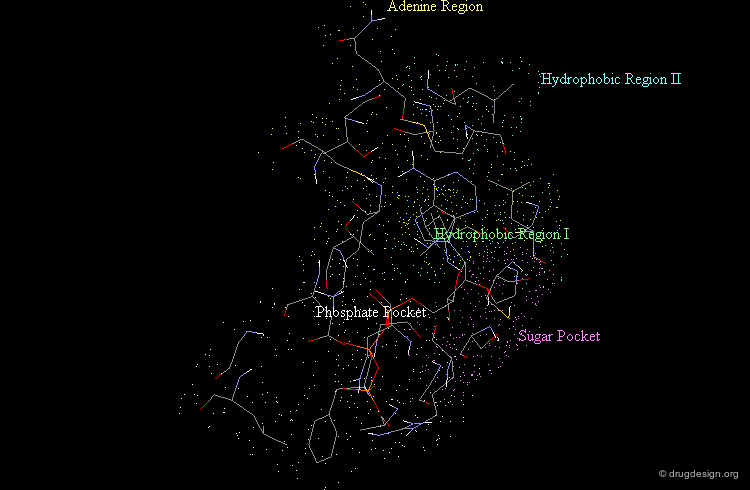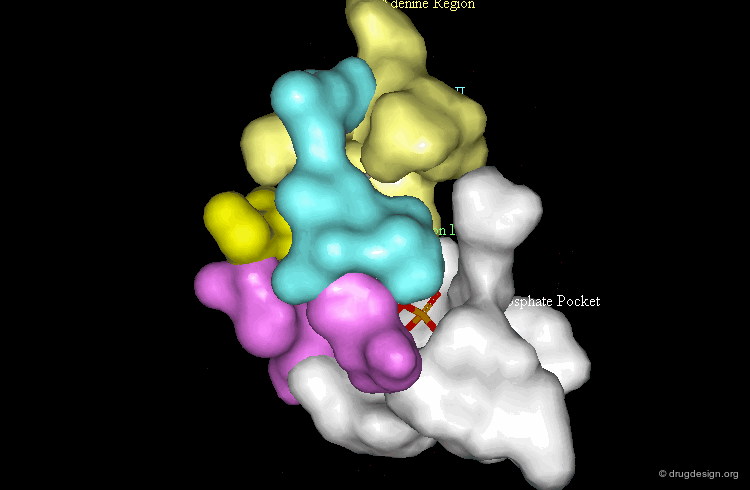
Morphology of the Active Site of a Protein Kinase¶
For example the catalytic site of the protein kinases can be well documented based on the binding of ATP, the common cofactor of all these enzymes. One could distinguish the adenine anchorage, the sugar and the phosphate pockets, as well as region I and II whose nature depends on the kinase considered. In the example shown here they both correspond to hydrophobic pockets.

Complexes with Ligands¶
For data consisting of a complex of the macromolecule with a ligand, the detailed interactions of the ligand are analyzed for good fits, conformational changes, and potential bump areas.

Forces That Contribute to the Binding¶
Molecular interactions are controlled by different driving forces. The principal forces involved upon the binding of a ligand to a biological system are hydrophobic interactions, hydrogen bonding and electrostatic forces. It has been proposed that the hydrophobic component (which is non-directional) contributes primarily to affinity whereas hydrogen bonding and electrostatic interactions, because of their highly directional nature, contribute principally to specificity.

The Molecular Recognition Process¶
The main forces involved in molecular recognition are the following: electrostatic, hydrogen bonding and hydrophobic.

Electrostatic¶
At long-distances, the electrostatic forces are dominant and control the preliminary approach of the ligand molecule to the active site.

Hydrogen Bonding¶
At medium-range distances, in addition to the electrostatic forces, hydrogen bonds begin to be formed. They start to establish anchorage points for positioning the ligand more precisely.

Hydrophobic¶
At shorter distances, and this is the last step of the process, hydrophobic forces become the dominant driving force.

Hydrophobic Interactions¶
In aqueous media hydrophobic groups tend to cluster in order to reduce non-polar surface areas exposed to water. The following table gives a list of the 9 hydrophobic residues out of the 20 naturally occurring amino acid residues present in protein structures.
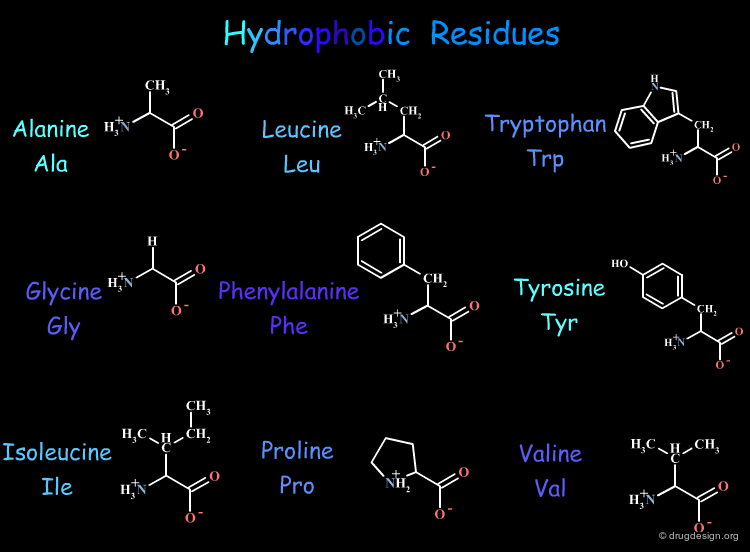
articles
Hydrophobic Effects. Opinion and Facts Blokzijl W and Engberts JBFN Angew. Chem. Int. Ed. Engl. 32 1993
X-ray Structures of Small Ligand-FKBP Complexes Provide an Estimate for Hydrophobic Interaction Energies Burkhard P, Taylor P and Walkinshaw MD J. Mol. Biol. 295 2000
Aromatic-Aromatic Interaction: a Mechanism of Protein Structure Stabilization Burley SK and Petsko GA Science 229 1985
Hydrophobic Bonding and Accessible Surface Area in Proteins Chothia C Nature 248 1974
3D Molecular Lipophilicity Potential Profiles: A New Tool in Molecular Modeling Furet P, Sele A and Cohen NC J. Mol. Graph 6 1988
CH/pi Interaction in the Packing of the Adenine Ring in Protein Structures Chakrabarti P and Samanta U J. Mol. Biol. 251 1995
Consider Hydrophobic Interactions¶
The driving force of hydrophobic interactions between a ligand and a receptor is to reduce the exposure of hydrophobic moieties of the ligand to water. In addition, there is a favorable interaction between lipophilic groups in contact (mainly dispersion interactions). High occupancy of hydrophobic pockets of the receptor by a ligand leads to favorable van der Waals attractions, which results in increasing the binding affinity. Therefore, the more empty space in the binding site, the weaker the affinity of the ligand. Note that, although the hydrophobic interactions are weak, there are a lot of them, so the effect can be substantial.
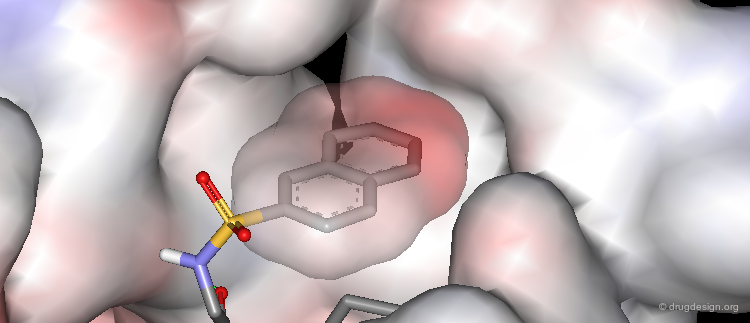
articles
Hydrophobic Effects. Opinion and Facts Blokzijl W and Engberts JBFN Angew. Chem. Int. Ed. Engl. 32 1993
X-ray Structures of Small Ligand-FKBP Complexes Provide an Estimate for Hydrophobic Interaction Energies Burkhard P, Taylor P and Walkinshaw MD J. Mol. Biol. 295 2000
Aromatic-Aromatic Interaction: a Mechanism of Protein Structure Stabilization Burley SK and Petsko GA Science 229 1985
Hydrophobic Bonding and Accessible Surface Area in Proteins Chothia C Nature 248 1974
3D Molecular Lipophilicity Potential Profiles: A New Tool in Molecular Modeling Furet P, Sele A and Cohen NC J. Mol. Graph 6 1988
CH/pi Interaction in the Packing of the Adenine Ring in Protein Structures Chakrabarti P and Samanta U J. Mol. Biol. 251 1995
Elementary Hydrophobic Interactions¶
Examples of hydrophobic interactions found in X-ray complexes between various amino acids and ligands are presented here.

articles
Hydrophobic Effects. Opinion and Facts Blokzijl W and Engberts JBFN Angew. Chem. Int. Ed. Engl. 32 1993
X-ray Structures of Small Ligand-FKBP Complexes Provide an Estimate for Hydrophobic Interaction Energies Burkhard P, Taylor P and Walkinshaw MD J. Mol. Biol. 295 2000
Aromatic-Aromatic Interaction: a Mechanism of Protein Structure Stabilization Burley SK and Petsko GA Science 229 1985
Hydrophobic Bonding and Accessible Surface Area in Proteins Chothia C Nature 248 1974
3D Molecular Lipophilicity Potential Profiles: A New Tool in Molecular Modeling Furet P, Sele A and Cohen NC J. Mol. Graph 6 1988
CH/pi Interaction in the Packing of the Adenine Ring in Protein Structures Chakrabarti P and Samanta U J. Mol. Biol. 251 1995
Example of Hydrophobic Binding¶
An example that illustrates the importance of hydrophobic interactions is the design of thyroid hormone analogues. The thyroxine-prealbumin complex was used as a model for the interaction of thyroid hormones with the nuclear thyroid hormone receptor. It was observed that thyroxine fills effectively only five of the six existing pockets of prealbumin. In order to improve the hydrophobic contacts with the sixth pocket, the bromo atom was replaced by a fused phenyl ring (the naphthyl derivative). As expected this substitution led to an increase in binding affinity.
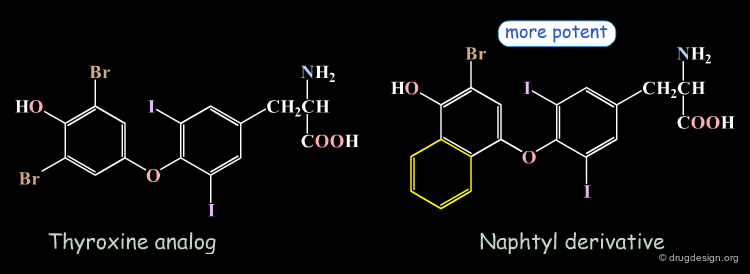
articles
Computer Graphics in Drug Design: Molecular Modeling of Thyroid Hormone-Prealbumin Interactions Blaney JM, Jorgensen EC, Connolly ML, Ferrin TE, Langridge R, Oatley SJ, Burridge JM and Blake CC J. Med. Chem. 25 1982
Strengthening Hydrophobic Interactions¶
It can be clearly seen that the naphthyl derivative fills the hydrophobic pocket better than the thyroxine-analog.

Hydrogen Bond Features¶
Hydrogen bonds are directional and contribute to the positioning of a molecular fragment in a precise orientation in the active site of a protein. Multiple hydrogen bonds can "zip" the ligand into place.

articles
Hydrogen Bonding in Globular Proteins Baker EN and Hubbard RE Prog. Biophys. Mol. Biol. 44 1984
The Role of Hydrogen Bonds in Protein Folding and Protein Association Ben-Naim A J. Phys. Chem 95 1991
Enthalpy of Hydrogen Bond Formation in a Protein-ligand Binding Reaction Connelly PR, Aldape RA, Bruzzese FJ, Chambers SP, Fitzgibbon MJ, Fleming MA, Itoh S, Livingston DJ, Navia MA, Thomson JA and Wilson KP Proc. Natl. Acad. Sci. U.S.A. 91 1994
The Hydrogen Bond in Molecular Recognition Fersht AR Trends. Biochem. Sci. 12 1987
Hydrogen Bond Stereochemistry in Protein Structure and Function Ippolito JA, Alexander RS and Christianson DW J. Mol. Biol. 215 1990
Aromatic Rings Act as Hydrogen Bond Acceptors Levitt M and Perutz MF J Mol Biol. 201 1988
The Nature of Intramolecular Hydrogen-Bonded and Non- Hydrogen-Bonded Conformations of Simple Di- and Triamides Novoa J and Whanghbo MH J. Am. Chem. Soc. 20 1991
Analysis of Hydrogen Bonding Strengh in Proteins Using Unnatural Amino Acids Thorson JS, Chapman E and Schultz PG J. Am. Chem. Soc. 117 1995
Proteins Capabilities in Hydrogen Bonding¶
A protein has many hydrogen bond donor and acceptor groups. In addition to the backbone peptidic moiety that can form hydrogen bonds with the C=O or the N-H, hydrogen bonds can be formed with the side chains of the residues.

articles
Hydrogen Bonding in Globular Proteins Baker EN and Hubbard RE Prog. Biophys. Mol. Biol. 44 1984
The Role of Hydrogen Bonds in Protein Folding and Protein Association Ben-Naim A J. Phys. Chem 95 1991
Enthalpy of Hydrogen Bond Formation in a Protein-ligand Binding Reaction Connelly PR, Aldape RA, Bruzzese FJ, Chambers SP, Fitzgibbon MJ, Fleming MA, Itoh S, Livingston DJ, Navia MA, Thomson JA and Wilson KP Proc. Natl. Acad. Sci. U.S.A. 91 1994
The Hydrogen Bond in Molecular Recognition Fersht AR Trends. Biochem. Sci. 12 1987
Hydrogen Bond Stereochemistry in Protein Structure and Function Ippolito JA, Alexander RS and Christianson DW J. Mol. Biol. 215 1990
Aromatic Rings Act as Hydrogen Bond Acceptors Levitt M and Perutz MF J Mol Biol. 201 1988
The Nature of Intramolecular Hydrogen-Bonded and Non- Hydrogen-Bonded Conformations of Simple Di- and Triamides Novoa J and Whanghbo MH J. Am. Chem. Soc. 20 1991
Analysis of Hydrogen Bonding Strengh in Proteins Using Unnatural Amino Acids Thorson JS, Chapman E and Schultz PG J. Am. Chem. Soc. 117 1995
Consider Hydrogen Bond Formations¶
Hydrogen bonds formed between ligand and receptor are at the expense of bonds broken between ligand and solvent and between receptor and solvent. As a result of the release of water molecules, the breaking of their hydrogen bonds and the formation of new hydrogen bonds with the ligand occurred.
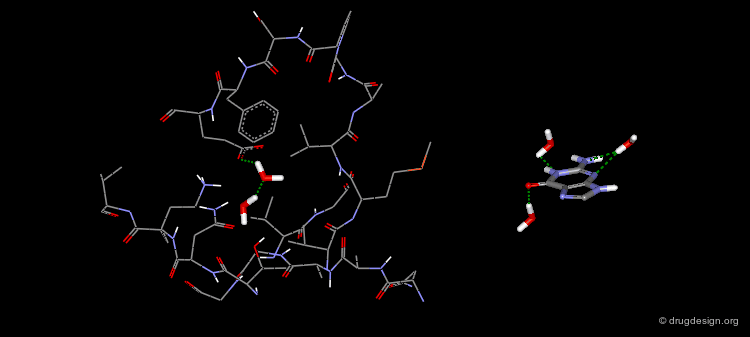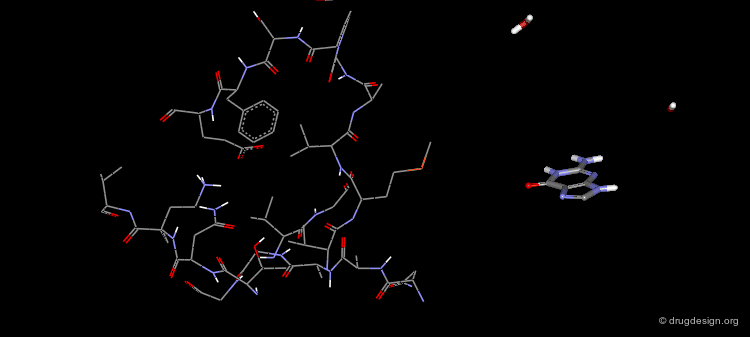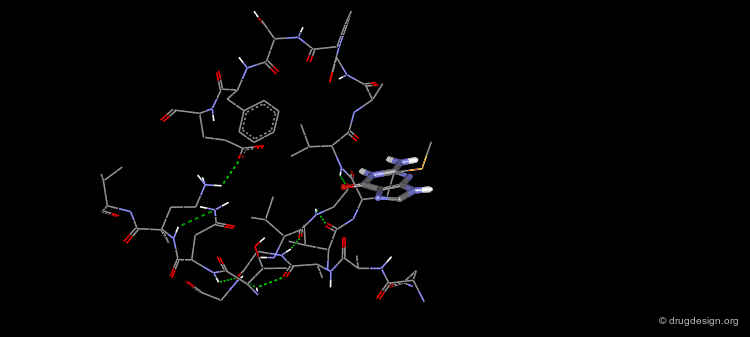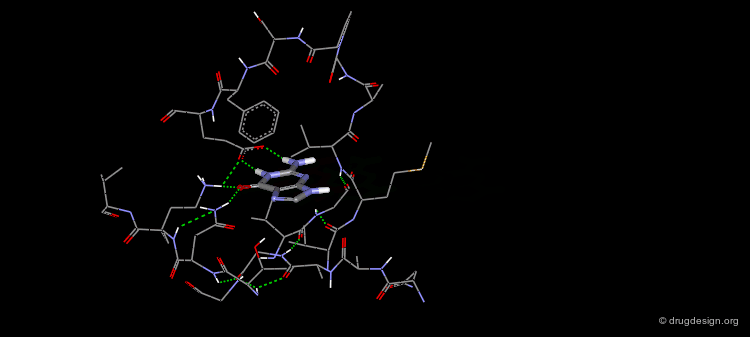
articles
Hydrogen Bonding in Globular Proteins Baker EN and Hubbard RE Prog. Biophys. Mol. Biol. 44 1984
The Role of Hydrogen Bonds in Protein Folding and Protein Association Ben-Naim A J. Phys. Chem 95 1991
Enthalpy of Hydrogen Bond Formation in a Protein-ligand Binding Reaction Connelly PR, Aldape RA, Bruzzese FJ, Chambers SP, Fitzgibbon MJ, Fleming MA, Itoh S, Livingston DJ, Navia MA, Thomson JA and Wilson KP Proc. Natl. Acad. Sci. U.S.A. 91 1994
The Hydrogen Bond in Molecular Recognition Fersht AR Trends. Biochem. Sci. 12 1987
Hydrogen Bond Stereochemistry in Protein Structure and Function Ippolito JA, Alexander RS and Christianson DW J. Mol. Biol. 215 1990
Aromatic Rings Act as Hydrogen Bond Acceptors Levitt M and Perutz MF J Mol Biol. 201 1988
The Nature of Intramolecular Hydrogen-Bonded and Non- Hydrogen-Bonded Conformations of Simple Di- and Triamides Novoa J and Whanghbo MH J. Am. Chem. Soc. 20 1991
Analysis of Hydrogen Bonding Strengh in Proteins Using Unnatural Amino Acids Thorson JS, Chapman E and Schultz PG J. Am. Chem. Soc. 117 1995
Elementary Hydrogen Bond Interactions¶
Examples of hydrogen bond interactions found in X-ray complexes between various amino acids and ligands are presented here.

articles
Hydrogen Bonding in Globular Proteins Baker EN and Hubbard RE Prog. Biophys. Mol. Biol. 44 1984
The Role of Hydrogen Bonds in Protein Folding and Protein Association Ben-Naim A J. Phys. Chem 95 1991
Enthalpy of Hydrogen Bond Formation in a Protein-ligand Binding Reaction Connelly PR, Aldape RA, Bruzzese FJ, Chambers SP, Fitzgibbon MJ, Fleming MA, Itoh S, Livingston DJ, Navia MA, Thomson JA and Wilson KP Proc. Natl. Acad. Sci. U.S.A. 91 1994
The Hydrogen Bond in Molecular Recognition Fersht AR Trends. Biochem. Sci. 12 1987
Hydrogen Bond Stereochemistry in Protein Structure and Function Ippolito JA, Alexander RS and Christianson DW J. Mol. Biol. 215 1990
Aromatic Rings Act as Hydrogen Bond Acceptors Levitt M and Perutz MF J Mol Biol. 201 1988
The Nature of Intramolecular Hydrogen-Bonded and Non- Hydrogen-Bonded Conformations of Simple Di- and Triamides Novoa J and Whanghbo MH J. Am. Chem. Soc. 20 1991
Analysis of Hydrogen Bonding Strengh in Proteins Using Unnatural Amino Acids Thorson JS, Chapman E and Schultz PG J. Am. Chem. Soc. 117 1995
Example of the Hydrogen Bond Binding¶
The binding of guanine analogs to the active site of purine nucleoside phosphorylase is an excellent example of the importance of the formation of hydrogen bonds. In this case the formation of one additional hydrogen bond increased the biological activity 100 times.


Electrostatic Interactions¶
Electrostatic interactions occur between polar or charged groups. The following residues contribute to the formation of electrostatic (coulombic or salt-bridge) interactions in proteins. Salt bridges are most stabilizing when they are somewhat shielded from solvent.

articles
Electrostatic Complementarity between proteins and ligands Chau PL and Dean PM J. Comput. Aided Mol. Des. 8 1994
Electrostatic Effects in Proteins Matthew JB Annu. Rev. Biophys. Biophys. Chem. 14 1985
Electrostatic Interactions in Macromolecules: Theory and Applications Sharp KA and Honig B Annu. Rev. Biophys. Biophys. Chem. 19 1990
Elementary Electrostatic Interactions¶
Examples of electrostatic interactions found in X-ray complexes between various amino acids and ligands are presented here.

articles
Electrostatic Complementarity between proteins and ligands Chau PL and Dean PM J. Comput. Aided Mol. Des. 8 1994
Electrostatic Effects in Proteins Matthew JB Annu. Rev. Biophys. Biophys. Chem. 14 1985
Electrostatic Interactions in Macromolecules: Theory and Applications Sharp KA and Honig B Annu. Rev. Biophys. Biophys. Chem. 19 1990
Strength of Electrostatic Interactions¶
Electrostatic interactions such as salt bridges and charge-assisted hydrogen bonds appear to be significantly stronger than neutral hydrogen bonds (in the range of -12 and -25 kJ/mol). This interaction energy is moderated by protonic solvents.

articles
Electrostatic Complementarity between proteins and ligands Chau PL and Dean PM J. Comput. Aided Mol. Des. 8 1994
Electrostatic Effects in Proteins Matthew JB Annu. Rev. Biophys. Biophys. Chem. 14 1985
Electrostatic Interactions in Macromolecules: Theory and Applications Sharp KA and Honig B Annu. Rev. Biophys. Biophys. Chem. 19 1990
Example of Electrostatic Interactions¶
Replacement of the hydroxyl group by an amino substituent resulted in a 400 times more potent neuraminidase (NA) inhibitor.

articles
Influenza Neuraminidase Inhibitors Possessing a Novel Hydrophobic Interaction in the Enzyme Active Site: Design, Synthesis, and Structural Analysis of Carboxylic Sialic Acid Analogues with Potent Anti-Influenza Activity. C.U. Kim et al. J.Am.Chem.Soc. 119 1997
Increase of Potency by the Formation of a Salt Bridge¶
The increase of potency is due to the formation of a salt bridge between the amino group and the carboxylate of Glu-119.
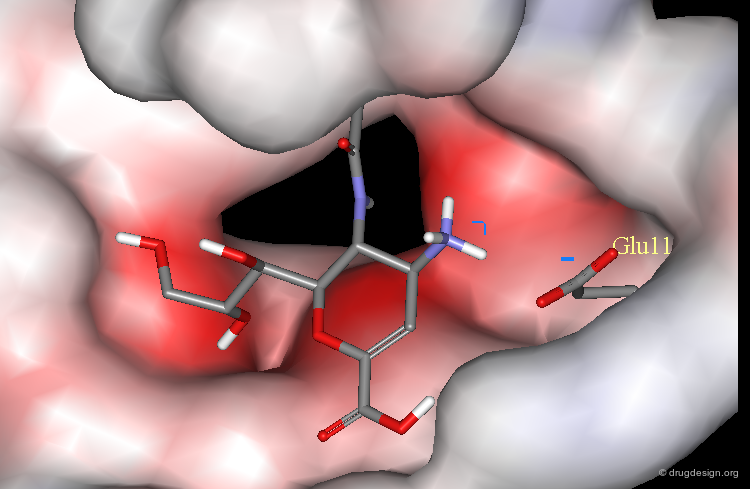
OH Analog Much Less Potent¶
The OH analog is 400 times less potent.
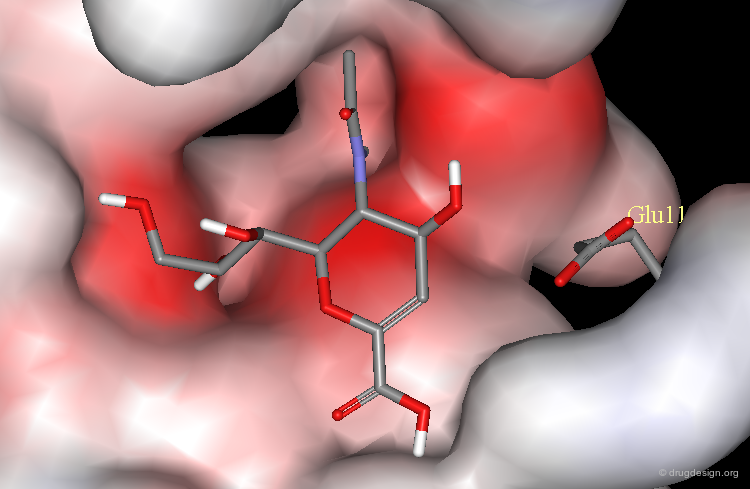
Example of Tight Interactions¶
An Example of a Tight Ligand-Receptor Interaction¶
Biotin is an enzyme cofactor which functions as a carrier of activated carbon dioxide. One of the tightest binding ligand-receptor pairs known is the complex between biotin and streptavidin (Ki = 0.1 pM). This tight binding is a consequence of high degree of shape and electrostatic complementarity between the ligand and the receptor. In addition, as a result of the binding of the ligand to the receptor, five ordered water molecules from the binding pocket of apo streptavidin are released.
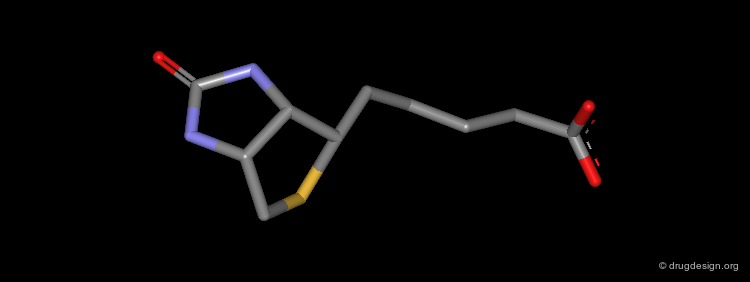

The X-ray Structure of the Biotin/Streptavidin¶
The crystal structure of the complex Biotin/Streptavidin has been resolved and is visualized here with some details.




The Binding Mode of Biotin with Streptavidin (1/4)¶
The stabilization of the complex is a result of the favorable interactions between a ligand and a receptor.

The Binding Mode of Biotin with Streptavidin (2/4)¶
2¶
The structure of this complex reveals a very complimentary steric fit between the ligand and the protein. Most of the accessible solvent surface of biotin is buried within streptavidin. The only exposed solvent group of biotin is the carboxylate.

The Binding Mode of Biotin with Streptavidin (3/4)¶
In addition to steric complementarity, there exists a very extended hydrogen bond network:The urea carbonyl makes three hydrogen bonds to the side chains of Tyr43, Ser27 and Asn23 with an approximately tetrahedral geometry.The two urea NH's form hydrogen bonds with Ser45 and Asp128 and the sulfur atom with Thr90.

The Binding Mode of Biotin with Streptavidin (4/4)¶
The side-chain carboxylate of biotin forms a charge-assisted hydrogen bond with Asn49 and a hydrogen bond with Ser88.

Receptor & Ligand Flexibility¶
Flexibility of the Receptor¶
A receptor possesses very flexible structural elements. The following is an example of the HIV-1 protease. X-ray crystallographic studies have shown that the tips of the flaps of this enzyme are capable of undergoing a 7 Å movement between the free and the complex enzyme.

articles
Specificity of Ligand Binding in a Buried Nonpolar Cavity of T4 Lysozyme: Linkage of Dynamics and Structural Plasticity Morton A and Matthews BW Biochemistry 34 1995
Protein Conformational Substates from X-ray Crystallography Rejto PA and Freer ST Prog. Biophys. Mol. Biol. 66 1996
The Conformation of the DNA Double Helix in the Crystal is Dependent on its Environment Shakked Z, Guerstein-Guzikevich, Eisenstein M, Frolow F and Rabinovich D Nature 342 1989
NMR Relaxation Studies of the Role of Conformational Entropy in Protein Stability and Ligand Binding Stone MJ Acc Chem Res. 34 2001
Flexibility of The Ligand¶
The flexibility of a ligand has direct consequences on the binding capabilities of the molecule and on the potency of the biological action. To understand ligand interactions, the bioactive conformation of the ligand and its probability of existence are of central importance. Making a flexible molecule rigid is a common strategy used to discover potent analogs.

Entropic Effects¶
From an entropic viewpoint, many aspects of complex formation are unfavorable as they result in the loss of conformational degrees of freedom for both the protein and the ligand. Therefore, highly favorable enthalpic contacts between the protein and the ligand must compensate for this entropic loss. Moreover, the more rigid the ligand is, the less rotational entropy is lost upon binding, giving higher affinity.

articles
Structure-Based Design of a Cyclophilin-Calcineurin Bridging Ligand Alberg DG and Schreiber SL Science 262 1993
Rationally Designed Dipeptoid Analogues of CCK. A Free-Wilson/Fijita-Ban Analysis of Some α-Methyltryptophan Derivatives as CCK-B Antagonists Higginbottom M, Kneen C and Ratcliffe GS J. Med. Chem. 35 1992
Design, Synthesis and Biological Activity of Novel Rigid Amidino-phenylalanine Derivatives as Inhibitors of Thrombin Mack H, Pfeiffer T, Hornberger W, Bohm HJ and Hoffken HW J. Enzyme. Inhib. 9 1995
Structure-Based Design of an Inhibitor of the Zinc Peptidase Thermolysin Morgan BP, Bartlett PA, Holland DR and Matthews BW J. Am. Chem. Soc. 116 1994
Role of the Solvent¶
Solvation and Desolvation¶
The binding of a ligand to a protein is a complex process in which the interacting entities become partially desolvated. This subtle thermodynamically driven chain of events leads to the formation of favorable interactions between the ligand and the protein. For example upon binding, hydrophobic moieties of the ligand enter into hydrophobic pockets of the receptor to minimize the contact with water.
The Role of the Solvent¶
The aqueous solvent plays a crucial role in determining the conformational preferences and interaction forces of ligands and receptors. In particular, the hydrophobic effect (also called hydrophobic collapse) is caused by the unfavorable interaction of non-polar groups with water molecules, they tend to cluster together to exclude water and minimize the area of interaction with water. The interpretation of this phenomenon is that water-water interactions are the most favorable interactions and in effect, the water squeezes out the hydrophobes so it can interact with itself, leaving the hydrophobes to interact with themselves.

Relay with Water Molecules¶
A ligand can bind directly to a receptor or indirectly through water molecules that are located between the ligand and the protein, establishing a hydrogen-bond network between them. The following example illustrates the case of a ligand where one water molecule contributes to the binding of the ligand.

Relay with Several Water Molecules¶
In addition to one water molecule connecting the protein and the ligand (on the right side of the adenine nucleus), two other water molecules are also contributing to the binding of this ligand (on the left side of the view).

Relay with Water Molecule Having Four H-Bonds¶
This other example illustrates the binding of a peptide-based molecule to the HIV-1 protease through a water molecule. The water molecule accepts two hydrogen bonds and donates another two; the four hydrogen bonds are in a tetrahedral geometry.

Prediction of Binding Modes¶
Binding Modes Predicted by Analogy¶
When an X-ray structure of a complex is available for a given ligand, the design is normally conceived with the assumption that the binding mode of the analog will be similar to that of the reference molecule. This has proven to be valid in many cases.

articles
Do structurally similar ligands bind in a similar fashion? Bostrom J, Hogner A, Schmitt S. J Med Chem. 49(23) 2006 10.1021/jm060167o
Inversion of Binding Modes¶
However experimental evidence indicates that this assumption may not be always valid. The following illustrates the case of molecules that were assumed to be of the same structure but experimental studies revealed unexpected binding modes. In many cases, the "unexpected" observation simply comes from insufficient development of the analyses.

Inverted Binding Mode of Olomoucine¶
ATP is the natural ligand of the protein kinases. A number of crystallographic studies of ATP and adenine-like analogs revealed the binding mode of the molecules of this family.

articles
Crystal Structure of Cyclin-Dependent Kinase 2 De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO and Kim SH Nature 363 1993
Multiple Modes of Ligand Recognition: Crystal Structures of Cyclin-dependent Protein Kinase 2 in Complex with ATP and Two Inhibitors, Olomoucine and Isopentenyladenine Schulze-Gahmen U, Brandsen J, Jones HD, Morgan DO, Meijer L, Vesely J and Kim SH Proteins 22 1995
Inhibition of Cyclin-dependent Kinases by Purine Analogues Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S and Meijer L Eur. J. Biochem. 224 1994
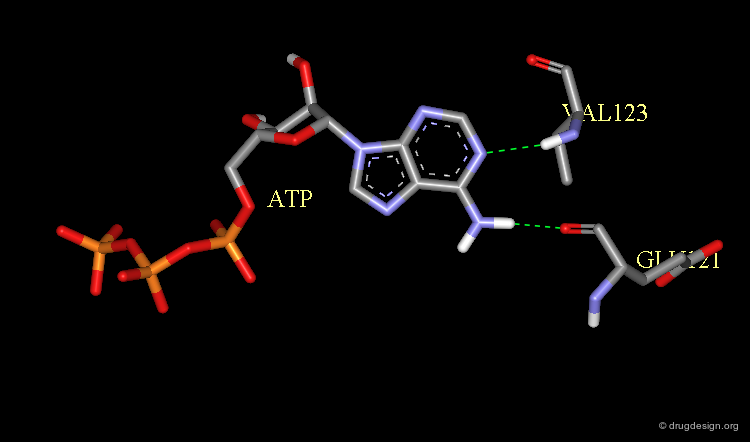
X-Ray Structure of ATP Bound to a Protein Kinase¶
The X-ray structure of the complex between ATP and the protein kinase CDK2 has been solved and confirms its now classical binding mode observed in all the kinases analyzed.
Intuitive 2D Alignment for Olomoucine¶
Olomoucine, a relatively selective inhibitor of the CDK2 kinase has an adenine-based scaffold that is also present in ATP. It was therefore anticipated that Olomoucine would have a comparable binding mode, corresponding to the following alignment.

Experimental Binding Mode of Olomoucine¶
The X-ray structure of the complex of Olomoucine with CDK2 reveals an entirely different binding mode and corresponds to an alignment with ATP that was unexpected.
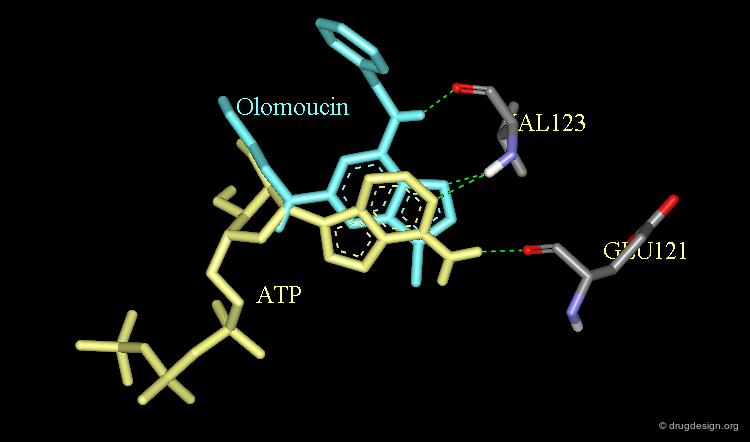
Origin of the Inverted Binding Mode of Olomoucine¶
Modeling analyses show that for steric reasons the Olomoucine molecule cannot conform to the binding of the authentic ligand ATP. The molecule binds to the catalytic site in a different orientation and another network of hydrogen bonds.

Inverted Binding Mode of Methotrexate¶
Dihydrofolate and methotrexate bind to the catalytic site of dihydrofolate reductase. Both have a rather long side chain (R) that is the same in the two molecules.

articles
Crystal Structure of Escherichia coli and Lactobacillus casei Dihydrofolate Reductase Refined at 1.7 A Resolution. I. General Features and Binding of Methotrexate Bolin JT, Filman DJ, Matthews DA, Hamlin RC and Kraut J J. Biol. Chem. 257 1982
Crystal Structures of Escherichia coli Dihydrofolate Reductase: the NADP+ Holoenzyme and the Folate. NADP+ Ternary Complex. Substrate Binding and a Model for the Transition State Bystroff C, Oatley SJ and Kraut J Biochemistry 29 1990
The Methotrexate Story: A Paradigm for Development of Cancer Chemotherapeutic Agents Huennekens FM Adv. Enzyme. Regul. 34 1994
Dihydrofolate Reductase: X-ray Structure of the Binary Complex with Methotrexate Matthews DA, Alden RA, Bolin JT, Freer ST, Hamlin R, Xuong N, Kraut J, Poe M, Williams M and Hoogsteen K Science 197 1977
Methotrexate Binding to Dihydrofolate Reductase Roberts GC, Feeney J, Birdsall B, Charlton P and Young D Nature 286 1980
Matching Chemistry and Shape in Molecular Docking Shoichet BK and Kuntz ID Protein. Eng. 6 1993
Intuitive 2D Alignment for Methotrexate¶
By simple observation of their chemical structures, it seems logical to imagine that the two molecules adopt similar binding modes, with the two structures aligned in the following way.

Experimental Binding Mode of Methotrexate¶
The X-ray structures of the complexes between the dihydrofolate and the methotrexate molecules indicate an entirely different binding mode for the two molecules, each molecule binding to the catalytic site upside down, relative to the other, while the methotrexate was found to be protonated.

Origin of the Inverted Binding Mode of Methotrexate¶
This can be understood by the better match of 3D distribution of the electrostatic properties of the two molecules by inverting one of them.

Binding Mode Predicted from SAR¶
When a sufficient number of analogs of a lead molecule are synthesized and tested, analysis of the resulting structure-activity relationships allows one to visualize the probable complementary receptor surface of this series of analogs.

Methods for Analyzing Binding¶
Analyzing Ligand-Receptor Binding¶
Computer graphics provide a powerful tool for analyzing the fit of a ligand into a complex macromolecular environment. Moreover computerized methods provide quantitative content to the detailed interaction energies involved in the binding of a molecule.

Ligand-Binding Predictions¶
The accurate and fast prediction of the binding energy for a protein-ligand complex is very important for the effective design and optimization of lead compounds. However, this is not an easy task. The binding of a ligand to a protein is a complex process.

Visual Analyses¶
Advanced computer graphics systems including stereo, real time rotations, volume and surface rendering provide in-depth analyses of the 3D stereo-chemical features of the active site of a protein and the favorable or unfavorable interaction of atoms or groups of atoms.

Docking Analyses¶
There are many forces that are involved in the intermolecular association or binding between two molecules. An energy-driven docking procedure that takes into account van der Waals, hydrogen bonding and electrostatic forces allows one to "dock" a ligand to a binding site by optimizing the interaction of the ligand and its receptor.

articles
A Good Ligands is Hard to Find: Automated Docking Methods Blaney JM and Dixon JS Perspect. Drug Discov. Design 1 1993
Docking Flexible Ligands to Macromolecular Receptors by Molecular Shape DesJarlais RL, Sheridan RP, Dixon JS, Kuntz ID and Venkataraghavan R J. Med. Chem. 29 1986
Recent Advances in Structure-based Rational Drug Design Gane PJ and Dean PM Curr. Opin. Struct. Biol. 10 2000
Docking Small-Molecule Ligands into Active Sites Jones G and Willett P Curr. Opin. Biotechnol. 6 1995
Theory of Macromolecule-Ligand Interactions Kollman PA Current Opinion in Structural Biology 4 1994
Geometric Approach to Macromolecule-Ligand Interactions Kuntz ID, Blaney JM, Oatley SJ, Langridge R and Ferrin TE J. Mol. Biol. 161 1982
Structure-based Strategies for Drug Design and Discovery Kuntz ID Science 257 1992
Ligand-Protein Docking and Rational Drug Design Lybrand TP Curr. Opin. Struct. Biol. 5 1995
Evaluation of Docking Functions for Protein-Ligand Docking Perez C and Ortiz AR J. Med. Chem. 44 2001
Flexible Docking and Design Rosenfeld R, Vajda S and DeLisi C Annu. Rev. Biophys. Biomol. Struct. 24 1995
Prediction of the Structure of a Receptor-Protein Complex Using a Binary Docking Method Stoddard BL and Koshland DE Jr Nature 358 1992
A Review of Protein-Small Molecule Docking Methods Taylor RD, Jewsbury PJ and Essex JW J. Comput. Aided. Mol. Des. 16 2002
Docking Ligands onto Binding Site Representations Derived from Proteins Built by Homology Modelling Schafferhans A and Klebe G J. Mol. Biol. 307 2001
Manual Docking with Computer Graphics¶
In manual methods ligands are constructed interactively in the active site of a macromolecular target by using computer graphic systems and then the complex is minimized. However, it is not easy to find the global-energy-minimum structure because many alternative conformations need to be explored to identify the preferred binding mode of a given molecule.

Automated Methods for Docking¶
Automated docking methods allow one to explore a number of possibilities in a systematic way. They allow a much more complete exploration of alternative binding modes as compared to interactive manual docking.

Calculation of Binding Energies¶
The accurate and fast prediction of the binding constant for a protein-ligand complex is very important for the effective design and optimization of lead compounds. Goodford proposed to evaluate the interaction energy on a grid of test points surrounding the target. This exploration reveals "hot areas" where favorable interactions can take place.

articles
Computational Methods to Predict Binding Free Energy in Ligand-Receptor Complexes Ajay MMA and Murcko MA J. Med. Chem. 38 1995
The Effect of Multiple Binding Modes on Empirical Modeling of Ligand Docking to Proteins Brem R and Dill KA Protein Sci. 8 1999
The Statistical-Thermodynamic basis for Computation of Binding Affinities: A Critical Review Gilson MK, Given JA, Bush BL and McCammon JA Biophys. J. 72 1997
A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules Goodford PJ J. Med. Chem. 28 1985
A Priori Prediction of Ligand Affinity by Energy Minimization Holloway MK Perspect. Drug Discov. Des. 9-11 1998
Computer Modeling of the Interactions of Complex Molecules Kollman PA and Merz KM Jr Acc. Chem. Res. 23 1990
Rapid Estimation of Relative Binding Affinities of Enzyme Inhibitors Reddy MR, Viswanadhan VN and Erion MD Perspect. Drug Discov. Des. 85 1998
book
Van Gunsteren WF
ESCOM 1989
Free Energy Perturbation Techniques¶
Free energy perturbation (FEP) calculation is an approach developed to calculate binding energies. Using a closed thermodynamic cycle (where the free energy between two states is independent of the path considered), molecular dynamics, and the experimental binding free energy of one inhibitor (ΔG1), FEP allows one to calculate free energy of new inhibitors (ΔG3). The method requires substantial computer time and thus it should not be used on a routine basis.

articles
Methodological Advances in Molecular Dynamics Simulations of Biological Systems Brooks CL 3rd Curr. Opin. Struct. Biol. 5 1995
Free Energy Calculations: Application to Chemical and Biochemical Phenomena Kollman P Chem. Rev. 93 1993
Free Energy from Simulations McCammon JA Curr. Opin. Struct. Biol. 1 1991
Free Energy Pertubation Simulation of the Inhibition of Thermolysin: Prediction of the Free Energy of Binding of a New Inhibitor Merz KM and Kollman PA J. Am. Chem. Soc. 111 1989
Absolute and Relative Binding Free Energy Calculations of the Interaction of Biotin and Its Analogs with Streptavidin Using Molecular Dynamics Free Energy Perturbation Approaches Miyamoto S and Kollman PA Proteins 16 1993
A New Method for Carrying Out Free Energy Perturbation Calculations: Dynamically Modified Windows Pearlman DA and Kollman PA J. Chem. Phys. 90 1989
Calculations of Relative Differences in Binding Free Energy of HIV-1 Protease Inhibitors: a Thermodynamic Perturbation Approach Reddy MR, Varney MD, Kalish V, Viswanadhan VN and Appelt K J. Med. Chem. 37 1994
Computer Simulation of Molecular Dynamics: Methodology, Applications and Perspectives in Chemistry Van Gunsteren WF and Berendsen HJC Angew. Chem. Int. Ed. Engl. 29 1990
Dynamics and Design of Enzyme Inhibitors Wong CF and McCammon JA J. Am. Chem. Soc. 108 1986
Energies from Force Field Calculations¶
The binding constant (K) of a ligand to its receptor is related to the ligand/receptor interaction energy (ΔH) via: ΔG = ΔH - TΔS = -RT lnK. If we assume the entropy change is the same for each molecule then we can write: ΔH = - RT lnK where the entalpy change on binding (interaction energy) is proportional to the log of the binding constant. The interaction energy can be calculated by force-field calculation as the sum of van der Waals and electrostatic interactions between the ligand and the receptor.

articles
Computer Modeling of the Interactions of Complex Molecules Kollman PA and Merz KM Jr Acc. Chem. Res 23 1990
Correlation with Biological Activities¶
This hypothesis has been shown to work in a handful of systems. In the case of HIV-1 protease inhibitors, we have X-ray data of co-crystallized ligand/receptor complexes of homologous series. A high correlation (r = 0.78) between the calculated interaction energy and biological activity (IC50) of a training set of 33 closely related HIV-1 protease inhibitors was observed. It was employed successfully to predict the potency of potential inhibitors such as the indane derivative, which is a precursor of indinavir.

Energies from Scoring Functions¶
Scoring functions are algorithms that assign a numerical score to a given ligand inside a receptor. The score should ideally give a measure of the ligand-protein binding constant. "Complementarity Score" are of moderate accuracy; the following illustrates the results obtained in the prediction of the binding constants of thermolysin inhibitors.

articles
The Development of a Simple Empirical Scoring Function to Estimate the Binding Constant for a Protein-Ligand Complex of Known Three-Dimensional Structure Bohm H-J J. Comput. Aided Mol. Design 8 1994
Knowledge-Based Scoring Function to Predict Protein-Ligand Iinteractions Gohlke H, Hendlich M and, Klebe G J. Mol. Biol. 295 2000
Evaluation of Docking/Scoring Approaches: A Comparative Study Based on MMP3 Inhibitors Ha S, Andreani R, Robbins A and Muegge I J Comput. Aided Mol. Des. 14 2000
Scoring Functions: a View from the Bench Tame JRH J. Comput. Aided. Mol. Des. 13 1999
Limitations of Scoring Functions¶
One of the limitations of scoring functions is that the conformational energy of the ligand is not taken into account. The predictions can be improved by calculating the difference of free energy between the docked conformation and the global minimum of the ligand.
Calculating Desolvation Energies¶
Accounting for solvent effects (e.g solvation/desolvation energies) is important for understanding of ligand-receptor interactions. Methods are now available to model the effects of solvents on both small and macromolecular solutes ranging from solvent continuum models to the incorporation of discrete solvent shells.
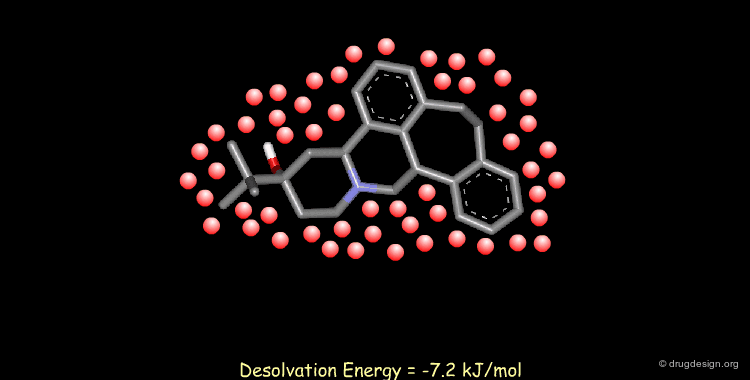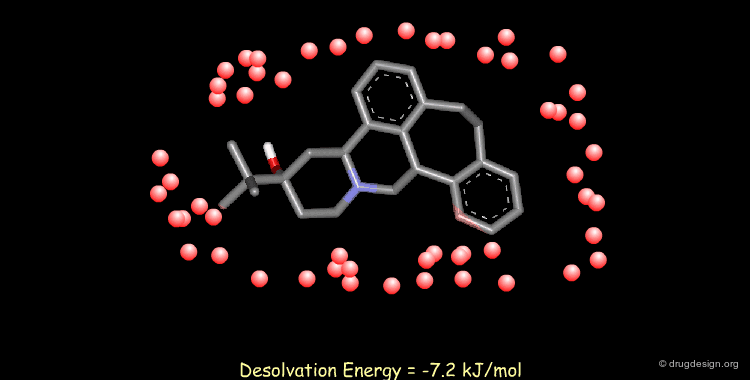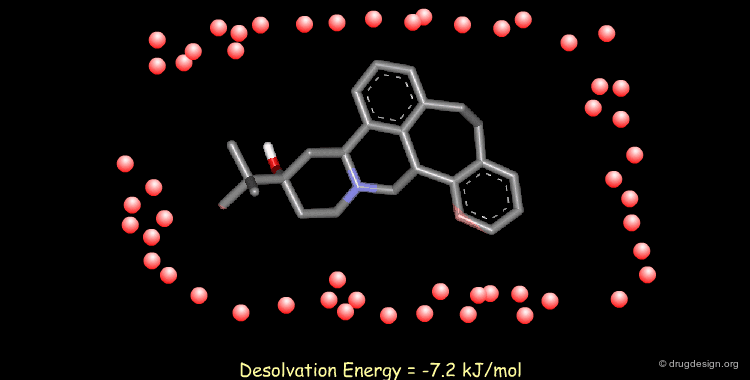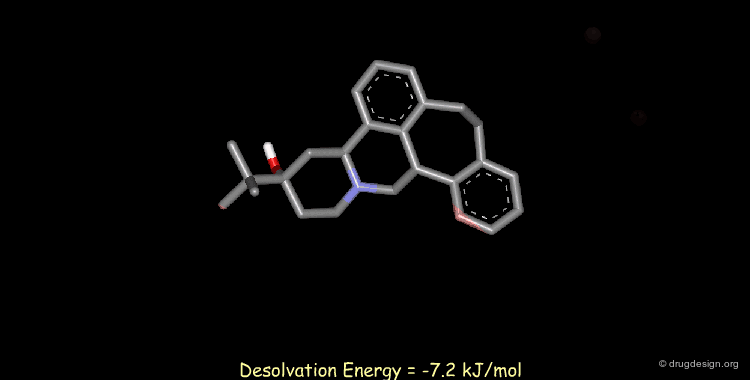
articles
An SCF Solvation Model for the Hydrophobic Effect and Absolute Free Energies of Aqueous Solvation Cramer JC and Truhlar DG Science 256 1992
Solvation Energy in Protein Folding and Binding Eisenberg D and McLachlan AD Nature 319 1986
Molecular Modeling in Solvent Milne GWA, Nicklaus MC and Hodoscek M J. Mol. Struct. 291 1993
Accessible Surface Areas as a Measure of the Thermodynamic Parameters of Hydration of Peptides Ooi T, Oobatake M, Nemethy G and Scheraga HA Proc. Natl. Acad. Sci. U.S.A. 84 1987
Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics Still WC, Tempczyk A, Hawley RC and Hendrickson T J. Am. Chem. Soc. 112 1990
Surface Area Included in Energy Refinement of Proteins, A Comparative Study on Atomic Solvation Parameters von Freyberg B, Richmond TJ and Braun W J. Mol. Biol. 233 1993
Atomic Solvation Parameters Applied to Molecular Dynamics of Proteins in Solution Wesson L and Eisenberg D Protein Science 1 1992
Empirical Solvation Models in the Context of Conformational Energy Searches: Application to Bovine Pancreatic Trypsin Inhibitor Williams RL, Vila J, Perrot G and Scheraga HA Proteins 14 1992
Inclusion of Solvation in Ligand Binding Free Energy Calculations Using the Generalized-Born Model Zou X, Sun Y and Kuntz ID J. Am. Chem. Soc. 121 1999
Copyright © 2024 drugdesign.org